

Comprehensive functional genomic resource and integrative model for the human brain
- PMID: 30545857
- PMCID: PMC6413328
- DOI: 10.1126/science.aat8464
Comprehensive functional genomic resource and integrative model for the human brain
Abstract
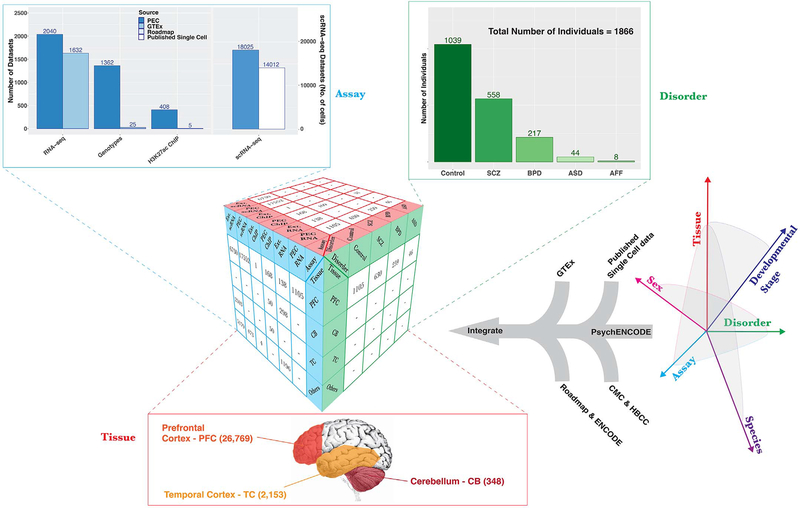
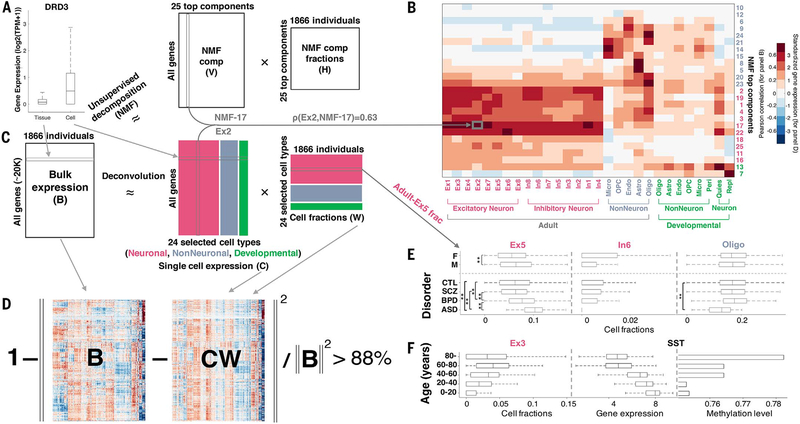
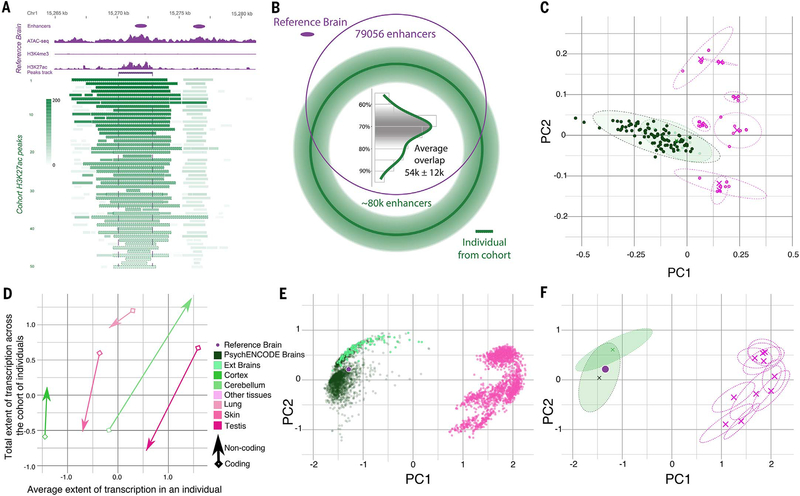
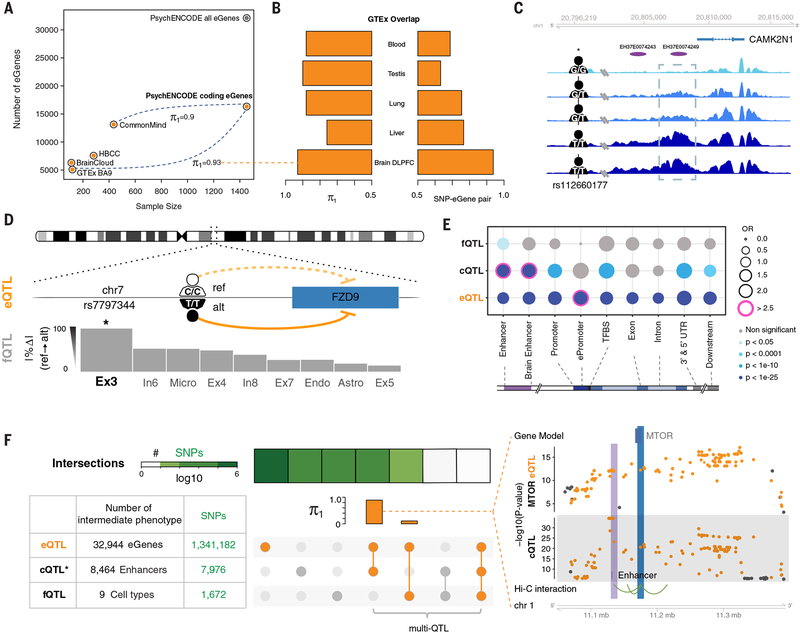
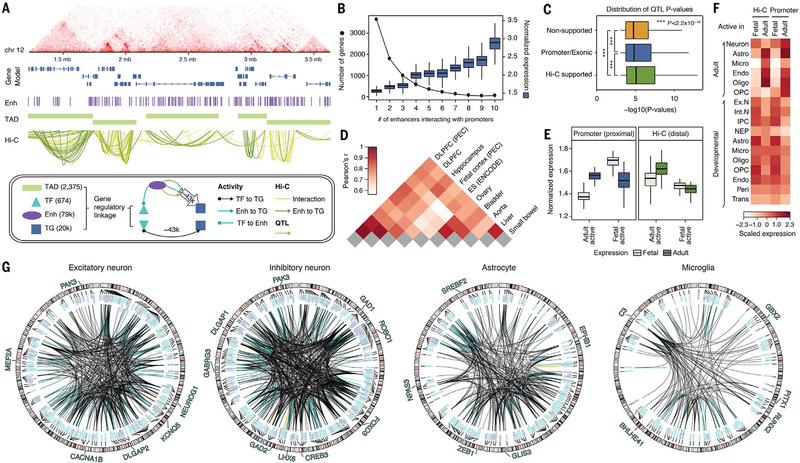
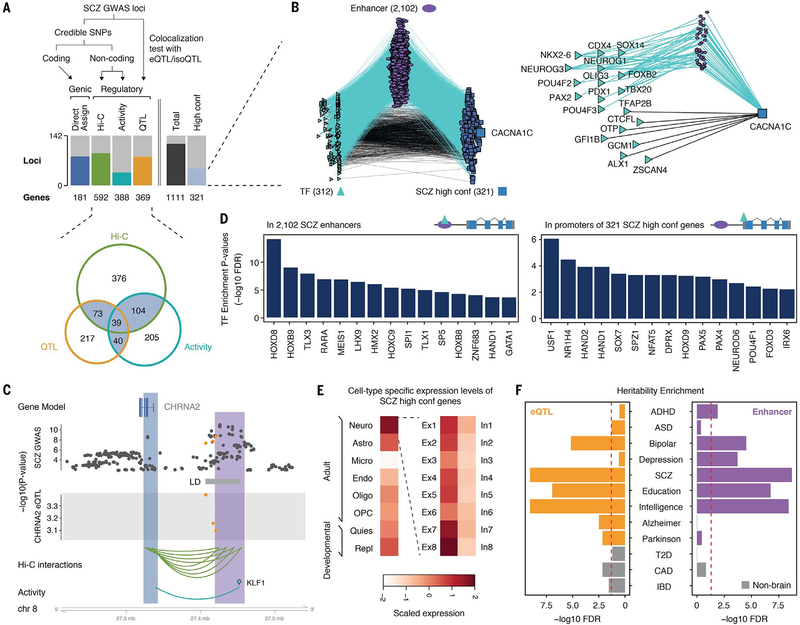
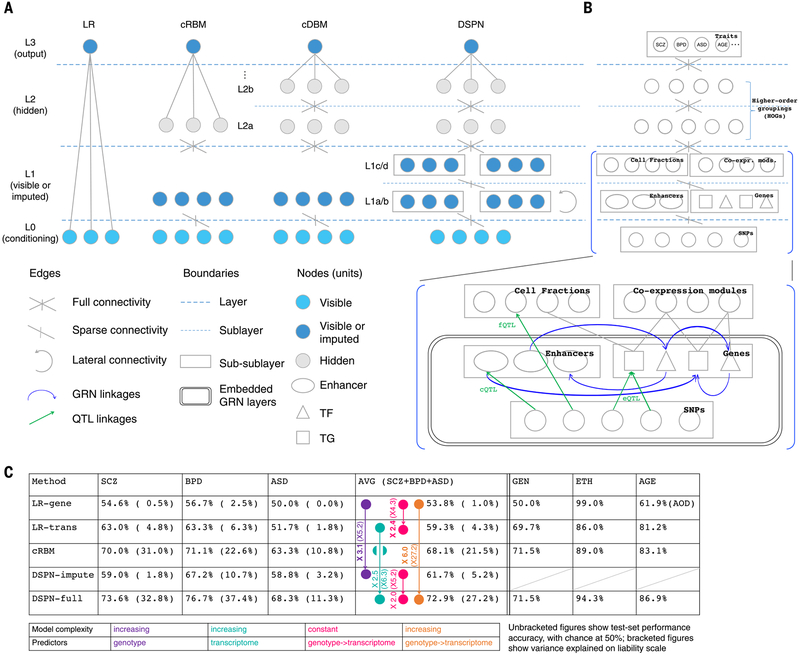
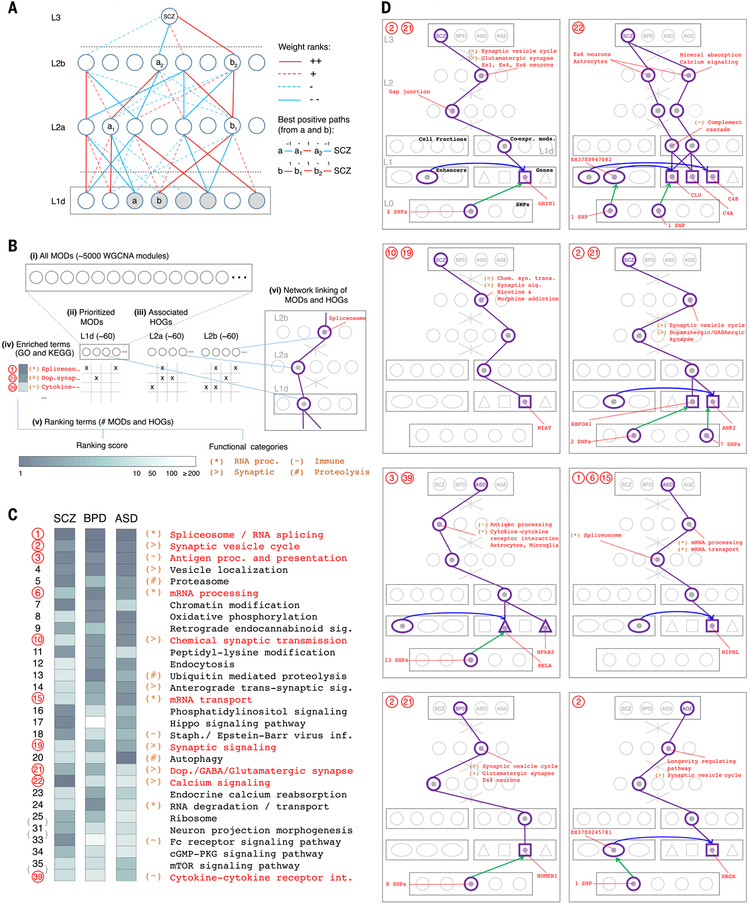
Despite progress in defining genetic risk for psychiatric disorders, their molecular mechanisms remain elusive. Addressing this, the PsychENCODE Consortium has generated a comprehensive online resource for the adult brain across 1866 individuals. The PsychENCODE resource contains ~79,000 brain-active enhancers, sets of Hi-C linkages, and topologically associating domains; single-cell expression profiles for many cell types; expression quantitative-trait loci (QTLs); and further QTLs associated with chromatin, splicing, and cell-type proportions. Integration shows that varying cell-type proportions largely account for the cross-population variation in expression (with >88% reconstruction accuracy). It also allows building of a gene regulatory network, linking genome-wide association study variants to genes (e.g., 321 for schizophrenia). We embed this network into an interpretable deep-learning model, which improves disease prediction by ~6-fold versus polygenic risk scores and identifies key genes and pathways in psychiatric disorders.
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Grants and funding
- R01 MH110928/MH/NIMH NIH HHS/United States
- R37 MH057881/MH/NIMH NIH HHS/United States
- R21 MH103877/MH/NIMH NIH HHS/United States
- U01 MH103365/MH/NIMH NIH HHS/United States
- R00 MH113823/MH/NIMH NIH HHS/United States
- R01 MH110927/MH/NIMH NIH HHS/United States
- R21 MH109956/MH/NIMH NIH HHS/United States
- U01 MH103392/MH/NIMH NIH HHS/United States
- R01 MH109677/MH/NIMH NIH HHS/United States
- U01 MH103346/MH/NIMH NIH HHS/United States
- R01 MH105472/MH/NIMH NIH HHS/United States
- P50 MH084053/MH/NIMH NIH HHS/United States
- R01 MH110920/MH/NIMH NIH HHS/United States
- P50 MH066392/MH/NIMH NIH HHS/United States
- R01 MH110926/MH/NIMH NIH HHS/United States
- R01 MH085542/MH/NIMH NIH HHS/United States
- P50 MH096891/MH/NIMH NIH HHS/United States
- R01 MH094714/MH/NIMH NIH HHS/United States
- R01 MH110905/MH/NIMH NIH HHS/United States
- R01 MH075916/MH/NIMH NIH HHS/United States
- S10 OD018521/OD/NIH HHS/United States
- K01 CA229995/CA/NCI NIH HHS/United States
- R01 MH111721/MH/NIMH NIH HHS/United States
- U01 MH103340/MH/NIMH NIH HHS/United States
- U01 MH103339/MH/NIMH NIH HHS/United States
- R01 MH097276/MH/NIMH NIH HHS/United States
- P01 AG002219/AG/NIA NIH HHS/United States
- R01 MH093725/MH/NIMH NIH HHS/United States
- U19 AI118610/AI/NIAID NIH HHS/United States
- P50 AG005138/AG/NIA NIH HHS/United States
- R21 MH102791/MH/NIMH NIH HHS/United States
- R01 MH105898/MH/NIMH NIH HHS/United States
- R21 MH105881/MH/NIMH NIH HHS/United States
- HHSN271201300031C/MH/NIMH NIH HHS/United States
- S10 OD030363/OD/NIH HHS/United States
- S10 OD018164/OD/NIH HHS/United States
- R01 MH110921/MH/NIMH NIH HHS/United States
- R21 MH105853/MH/NIMH NIH HHS/United States
- R01 MH109715/MH/NIMH NIH HHS/United States
- R01 MH080405/MH/NIMH NIH HHS/United States
- U01 MH116492/MH/NIMH NIH HHS/United States
- P50 MH106934/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
