

Genetics and genomics of pulmonary arterial hypertension
- PMID: 30545973
- PMCID: PMC6351337
- DOI: 10.1183/13993003.01899-2018
Genetics and genomics of pulmonary arterial hypertension
Abstract
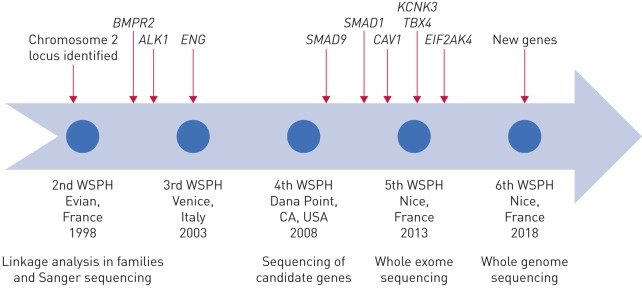
Since 2000 there have been major advances in our understanding of the genetic and genomics of pulmonary arterial hypertension (PAH), although there remains much to discover. Based on existing knowledge, around 25-30% of patients diagnosed with idiopathic PAH have an underlying Mendelian genetic cause for their condition and should be classified as heritable PAH (HPAH). Here, we summarise the known genetic and genomic drivers of PAH, the insights these provide into pathobiology, and the opportunities afforded for development of novel therapeutic approaches. In addition, factors determining the incomplete penetrance observed in HPAH are discussed. The currently available approaches to genetic testing and counselling, and the impact of a genetic diagnosis on clinical management of the patient with PAH, are presented. Advances in DNA sequencing technology are rapidly expanding our ability to undertake genomic studies at scale in large cohorts. In the future, such studies will provide a more complete picture of the genetic contribution to PAH and, potentially, a molecular classification of this disease.
Copyright ©ERS 2019.
Conflict of interest statement
Conflict of interest: N.W. Morrell reports grants and personal fees from Morphogen-IX, outside the submitted work. Conflict of interest: M.A. Aldred reports grants from the NIH, during the conduct of the study. Conflict of interest: W.K. Chung has nothing to disclose. Conflict of interest: C.G. Elliott reports personal fees for steering committee work from Bayer and Bellerophon, grants and personal fees for registry and data safety monitoring from Actelion, and was a consultant for end-point adjudication for Lung LLC, with fees paid to his employer (Intermountain Healthcare), outside the submitted work. Conflict of interest: W.C. Nichols has nothing to disclose. Conflict of interest: F. Soubrier has nothing to disclose. Conflict of interest: R.C. Trembath has nothing to disclose. Conflict of interest: J.E. Loyd has nothing to disclose.
Figures

Comment in
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous