

Tracking virus outbreaks in the twenty-first century
- PMID: 30546099
- PMCID: PMC6345516
- DOI: 10.1038/s41564-018-0296-2
Tracking virus outbreaks in the twenty-first century
Abstract
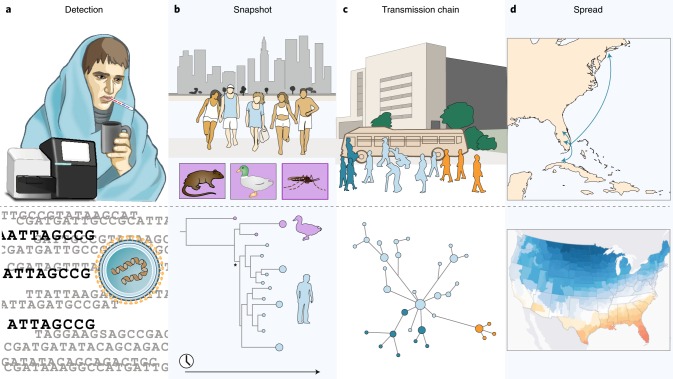
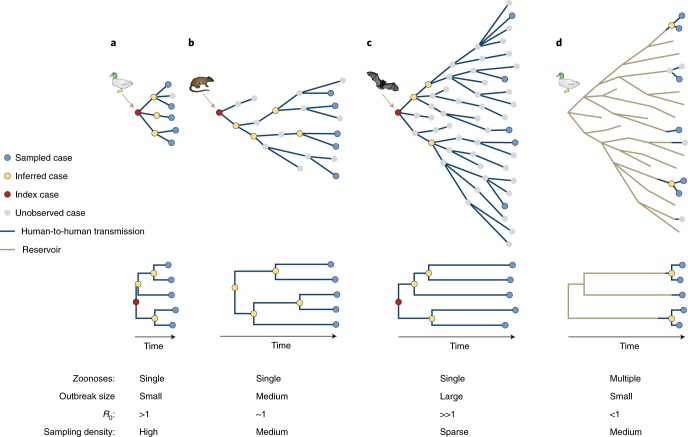
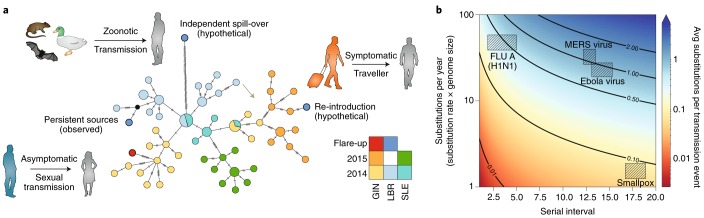
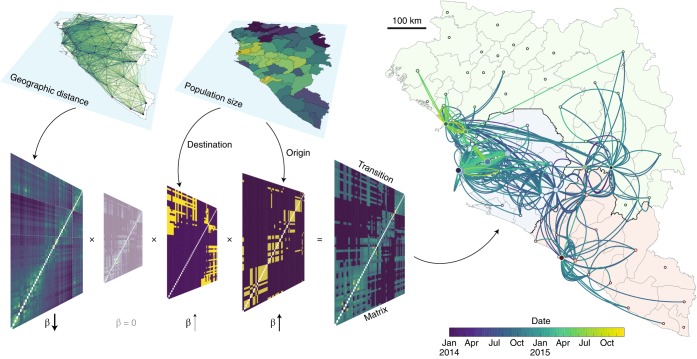
Emerging viruses have the potential to impose substantial mortality, morbidity and economic burdens on human populations. Tracking the spread of infectious diseases to assist in their control has traditionally relied on the analysis of case data gathered as the outbreak proceeds. Here, we describe how many of the key questions in infectious disease epidemiology, from the initial detection and characterization of outbreak viruses, to transmission chain tracking and outbreak mapping, can now be much more accurately addressed using recent advances in virus sequencing and phylogenetics. We highlight the utility of this approach with the hypothetical outbreak of an unknown pathogen, 'Disease X', suggested by the World Health Organization to be a potential cause of a future major epidemic. We also outline the requirements and challenges, including the need for flexible platforms that generate sequence data in real-time, and for these data to be shared as widely and openly as possible.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
