

Local genomic features predict the distinct and overlapping binding patterns of the bHLH-Zip family oncoproteins MITF and MYC-MAX
- PMID: 30548162
- PMCID: PMC6555698
- DOI: 10.1111/pcmr.12762
Local genomic features predict the distinct and overlapping binding patterns of the bHLH-Zip family oncoproteins MITF and MYC-MAX
Erratum in
-
Corrigendum.Pigment Cell Melanoma Res. 2020 May;33(3):520. doi: 10.1111/pcmr.12875. Pigment Cell Melanoma Res. 2020. PMID: 32319742 No abstract available.
Abstract
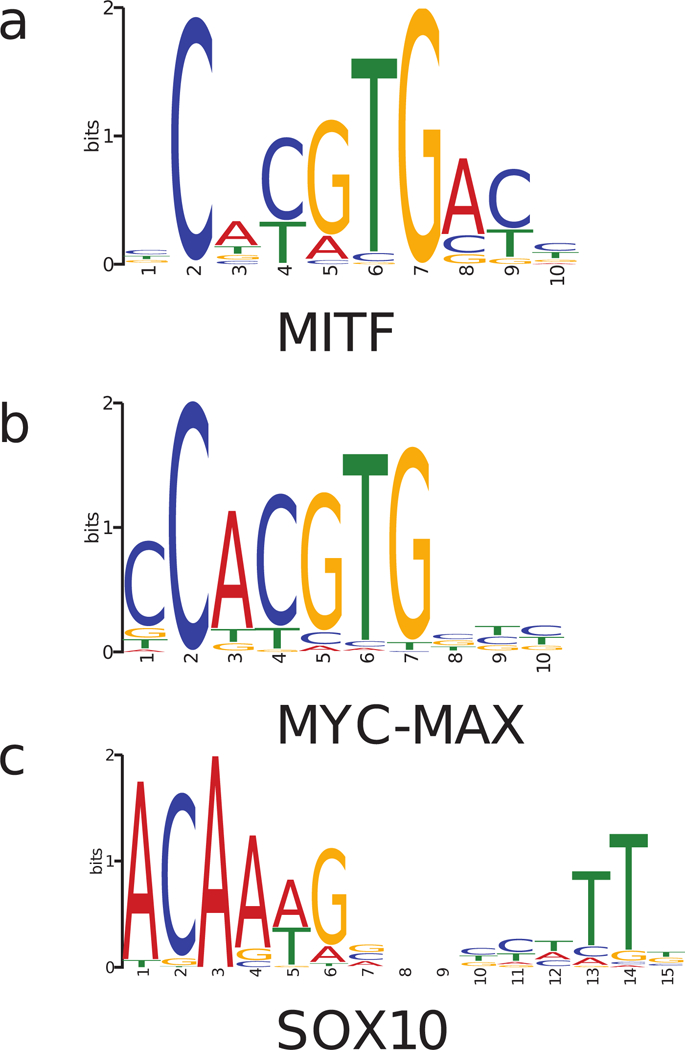
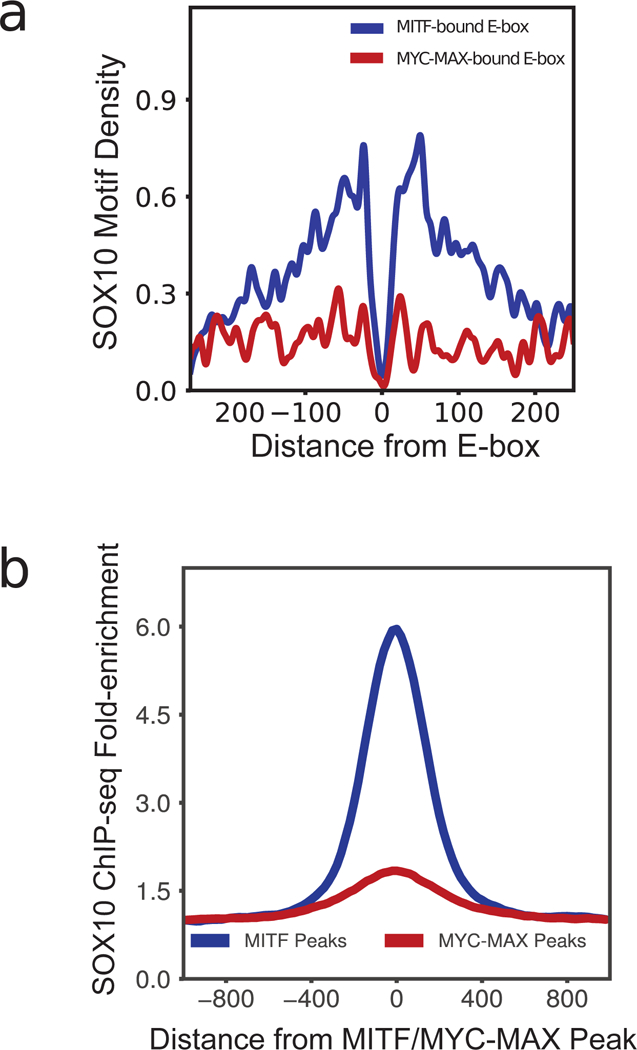
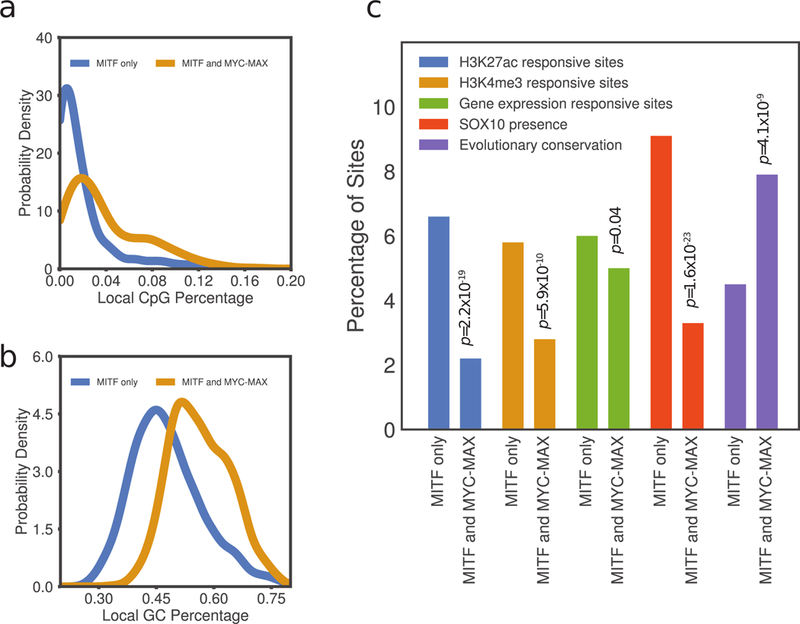
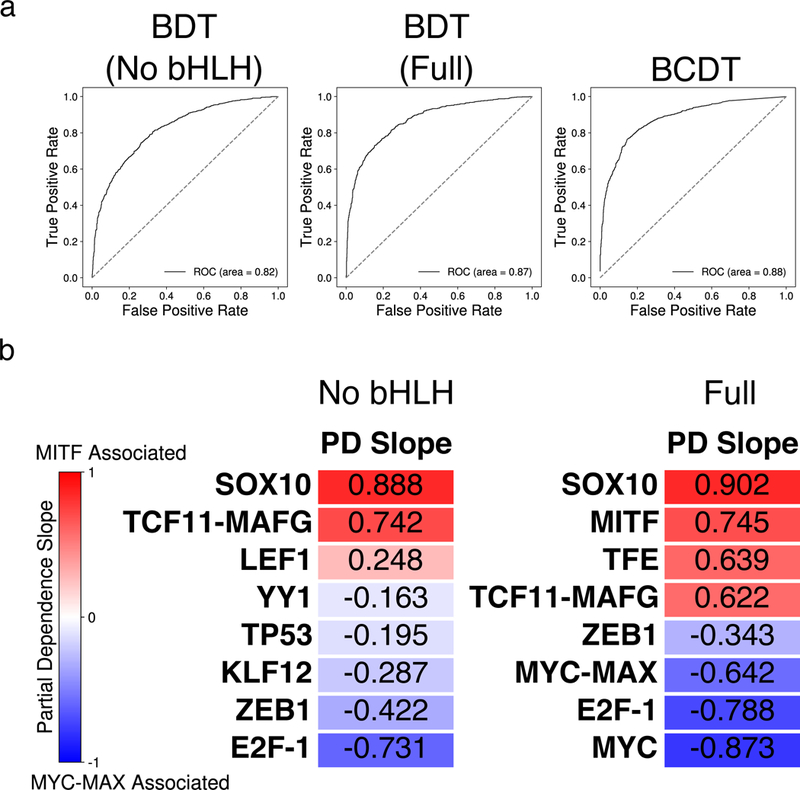
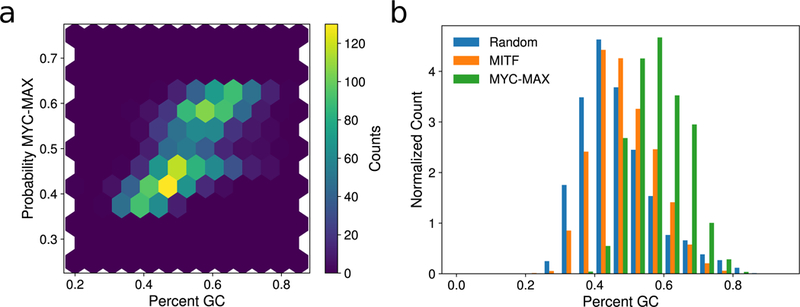
MITF and MYC are well-known oncoproteins and members of the basic helix-loop-helix leucine zipper (bHLH-Zip) family of transcription factors (TFs) recognizing hexamer E-box motifs. MITF and MYC not only share the core binding motif, but are also the two most highly expressed bHLH-Zip transcription factors in melanocytes, raising the possibility that they may compete for the same binding sites in select oncogenic targets. Mechanisms determining the distinct and potentially overlapping binding modes of these critical oncoproteins remain uncharacterized. We introduce computational predictive models using local sequence features, including a boosted convolutional decision tree framework, to distinguish MITF versus MYC-MAX binding sites with up to 80% accuracy genomewide. Select E-box locations that can be bound by both MITF and MYC-MAX form a separate class of MITF binding sites characterized by differential sequence content in the flanking region, diminished interaction with SOX10, higher evolutionary conservation, and less tissue-specific chromatin organization.
Keywords: MITF; MYC-MAX; boosted convolutional decision tree; cobinding factors; machine learning.
© 2018 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd.
Conflict of interest statement
Conflict of Interest
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
