

Integrating molecular networks with genetic variant interpretation for precision medicine
- PMID: 30548534
- PMCID: PMC6450710
- DOI: 10.1002/wsbm.1443
Integrating molecular networks with genetic variant interpretation for precision medicine
Abstract
More reliable and cheaper sequencing technologies have revealed the vast mutational landscapes characteristic of many phenotypes. The analysis of such genetic variants has led to successful identification of altered proteins underlying many Mendelian disorders. Nevertheless the simple one-variant one-phenotype model valid for many monogenic diseases does not capture the complexity of polygenic traits and disorders. Although experimental and computational approaches have improved detection of functionally deleterious variants and important interactions between gene products, the development of comprehensive models relating genotype and phenotypes remains a challenge in the field of genomic medicine. In this context, a new view of the pathologic state as significant perturbation of the network of interactions between biomolecules is crucial for the identification of biochemical pathways associated with complex phenotypes. Seminal studies in systems biology combined the analysis of genetic variation with protein-protein interaction networks to demonstrate that even as biological systems evolve to be robust to genetic variation, their topologies create disease vulnerabilities. More recent analyses model the impact of genetic variants as changes to the "wiring" of the interactome to better capture heterogeneity in genotype-phenotype relationships. These studies lay the foundation for using networks to predict variant effects at scale using machine-learning or algorithmic approaches. A wealth of databases and resources for the annotation of genotype-phenotype relationships have been developed to support developments in this area. This overview describes how study of the molecular interactome has generated insights linking the organization of biological systems to disease mechanism, and how this information can enable precision medicine. This article is categorized under: Translational, Genomic, and Systems Medicine > Translational Medicine Biological Mechanisms > Cell Signaling Models of Systems Properties and Processes > Mechanistic Models Analytical and Computational Methods > Computational Methods.
Keywords: disease mechanism; genetic disease; network analysis; variant interpretation.
© 2018 The Authors. WIREs Systems Biology and Medicine published by Wiley Periodicals, Inc.
Conflict of interest statement
The authors have declared no conflicts of interest for this article.
Figures
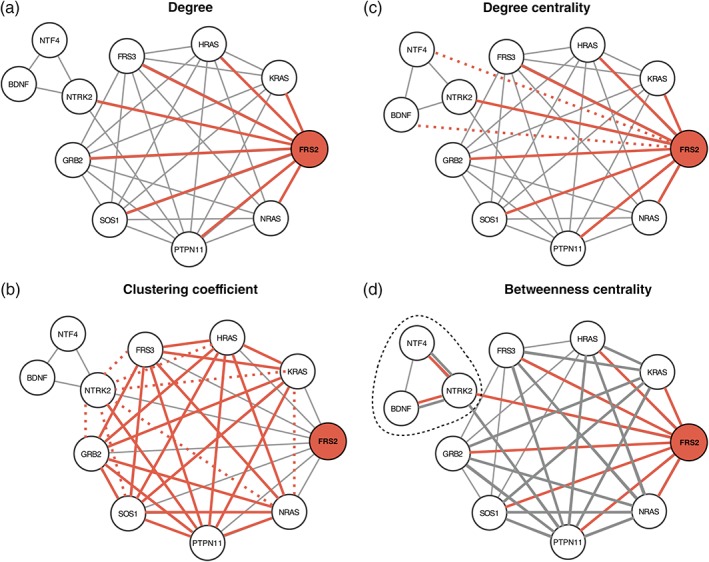
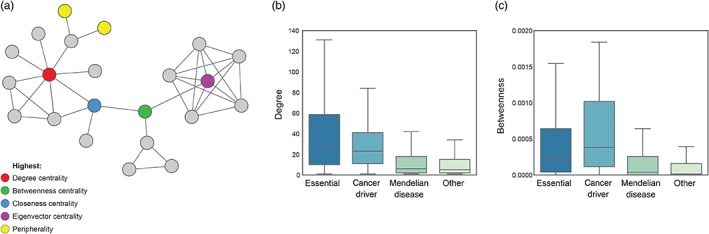
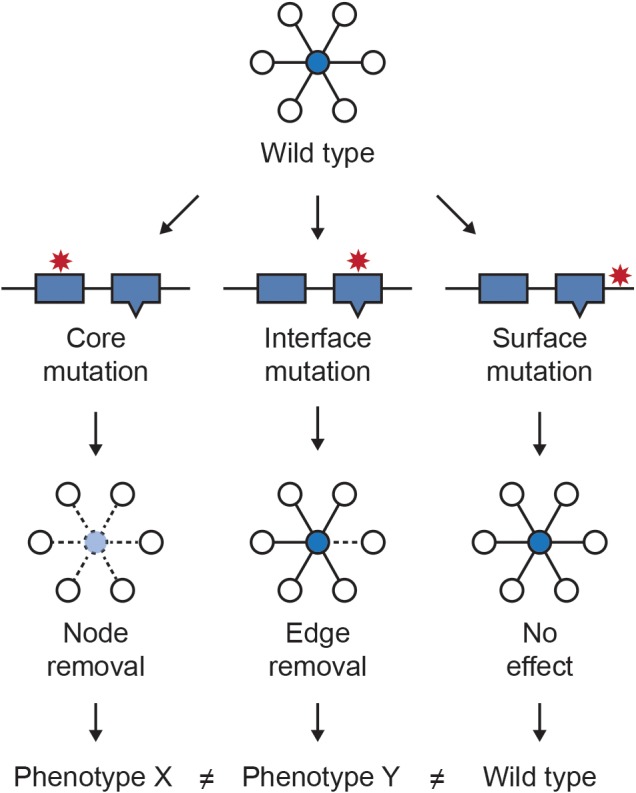
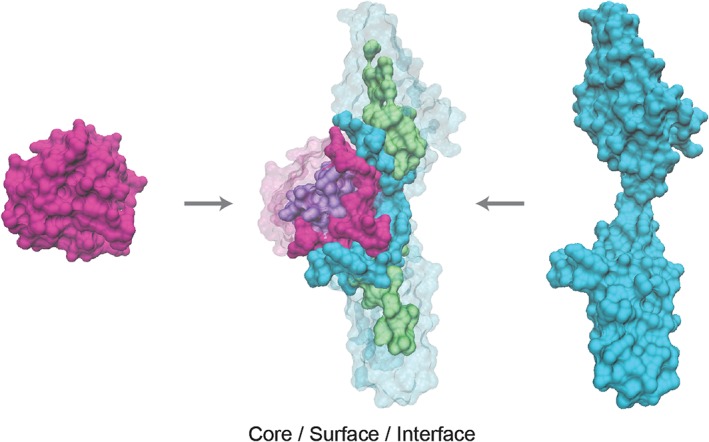
References
FURTHER READING
-
- Koller, D. , & Friedman, N. (2009). Probabilistic graphical models: Principles and techniques. Cambridge, MA: MIT Press.
References
-
- Abar, S. , Theodoropoulos, G. K. , Lemarinier, P. , & O'Hare, G. M. P. (2017). Agent based modelling and simulation tools: A review of the state‐of‐art software. Computer Science Review, 24, 13–33.
-
- Albert, R. (2005). Scale‐free networks in cell biology. Journal of Cell Science, 118, 4947–4957. - PubMed
-
- Albert, R. , Jeong, H. , & Barabasi, A. L. (2000). Error and attack tolerance of complex networks. Nature, 406, 378–382. - PubMed
