

Genotype, extrapyramidal features, and severity of variant ataxia-telangiectasia
- PMID: 30549301
- PMCID: PMC6590299
- DOI: 10.1002/ana.25394
Genotype, extrapyramidal features, and severity of variant ataxia-telangiectasia
Abstract
Objective: Variant ataxia-telangiectasia is caused by mutations that allow some retained ataxia telangiectasia-mutated (ATM) kinase activity. Here, we describe the clinical features of the largest established cohort of individuals with variant ataxia-telangiectasia and explore genotype-phenotype correlations.
Methods: Cross-sectional data were collected retrospectively. Patients were classified as variant ataxia-telangiectasia based on retained ATM kinase activity.
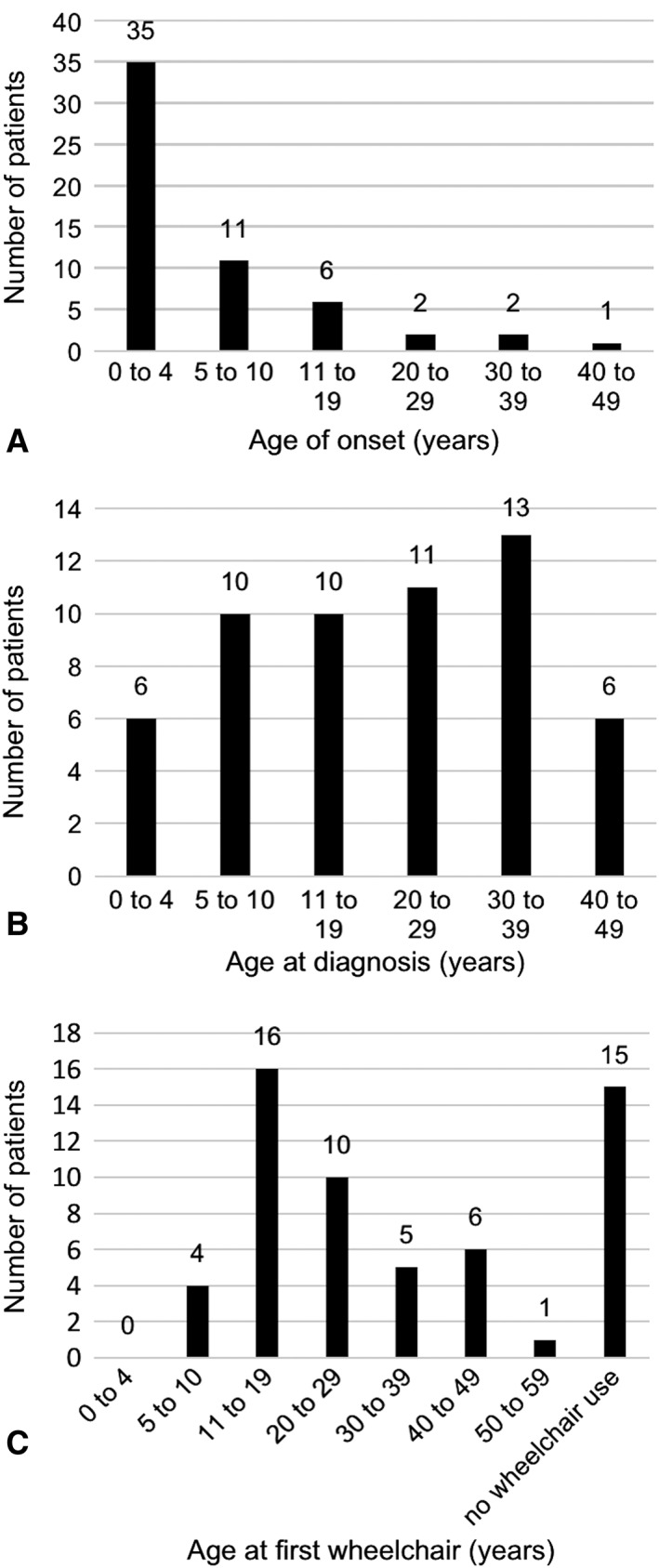
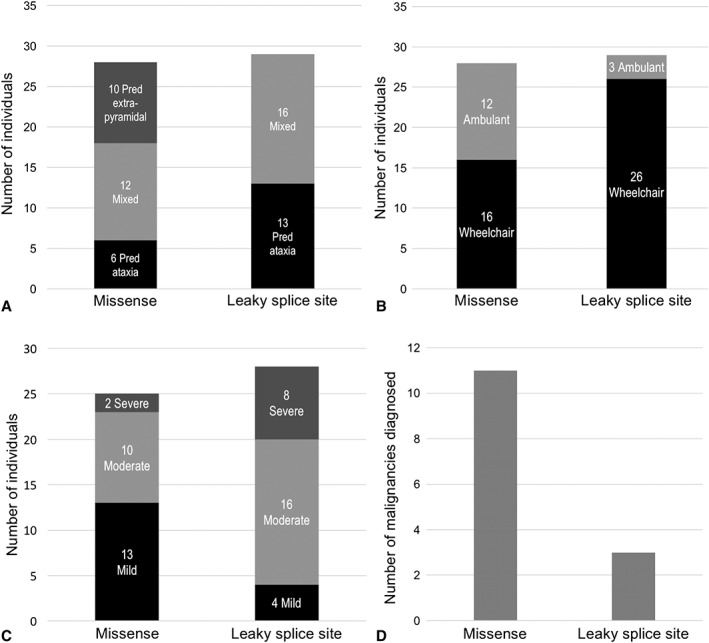
Results: The study includes 57 individuals. Mean age at assessment was 37.5 years. Most had their first symptoms by age 10 (81%). There was a diagnostic delay of more than 10 years in 68% and more than 20 years in one third of probands. Disease severity was mild in one third of patients, and 43% were still ambulant 20 years after disease onset. Only one third had predominant ataxia, and 18% had a pure extrapyramidal presentation. Individuals with extrapyramidal presentations had milder neurological disease severity. There were no significant respiratory or immunological complications, but 25% of individuals had a history of malignancy. Missense mutations were associated with milder neurological disease severity, but with a higher risk of malignancy, compared to leaky splice site mutations.
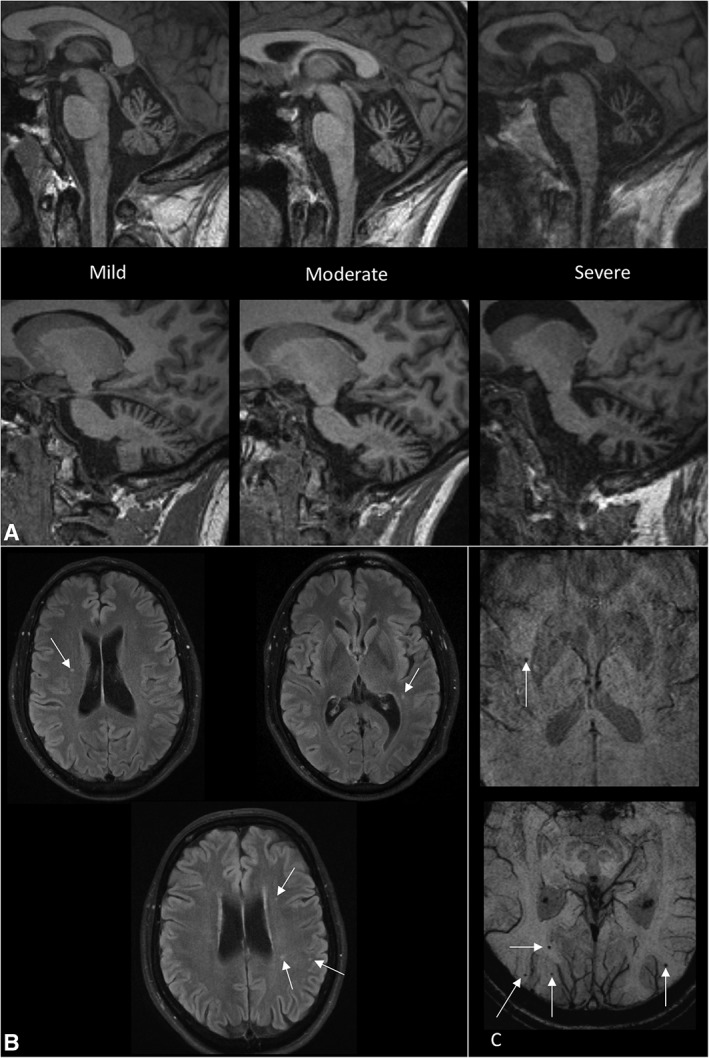
Interpretation: Individuals with variant ataxia-telangiectasia require malignancy surveillance and tailored management. However, our data suggest the condition may sometimes be mis- or underdiagnosed because of atypical features, including exclusive extrapyramidal symptoms, normal eye movements, and normal alpha-fetoprotein levels in some individuals. Missense mutations are associated with milder neurological presentations, but a particularly high malignancy risk, and it is important for clinicians to be aware of these phenotypes. ANN NEUROL 2019;85:170-180.
© 2018 The Authors. Annals of Neurology published by Wiley Periodicals, Inc. on behalf of American Neurological Association.
Conflict of interest statement
Nothing to report.
Figures
References
-
- Gatti RA, Berkel I, Boder E, et al. Localization of an ataxia‐telangiectasia gene to chromosome 11q22‐23. Nature 1988;336:577–580. - PubMed
-
- McKinnon PJ. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol 2012;7:303–321. - PubMed
-
- Woods CG, Bundey SE, Taylor AM. Unusual features in the inheritance of ataxia telangiectasia. Hum Genet 1990;84:555–562. - PubMed
-
- Verhagen MM, Last JI, Hogervorst FB, et al. Presence of ATM protein and residual kinase activity correlates with the phenotype in ataxia‐telangiectasia: a genotype‐phenotype study. Hum Mutat 2012;33:561–571. - PubMed
-
- Sedgwick RP, Boder E. Ataxia telangiectasia In: Vinken PJ, Bruyn SW, eds. Handbook of Clinical Neurology, Hereditary Neuropathies and Spino‐cerebellar Atrophies. New York, NY: Elsevier Science; 1991:347–393.
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
