

Intrinsic Dynamics of a Human Gene Reveal the Basis of Expression Heterogeneity
- PMID: 30554876
- PMCID: PMC6331006
- DOI: 10.1016/j.cell.2018.11.026
Intrinsic Dynamics of a Human Gene Reveal the Basis of Expression Heterogeneity
Abstract
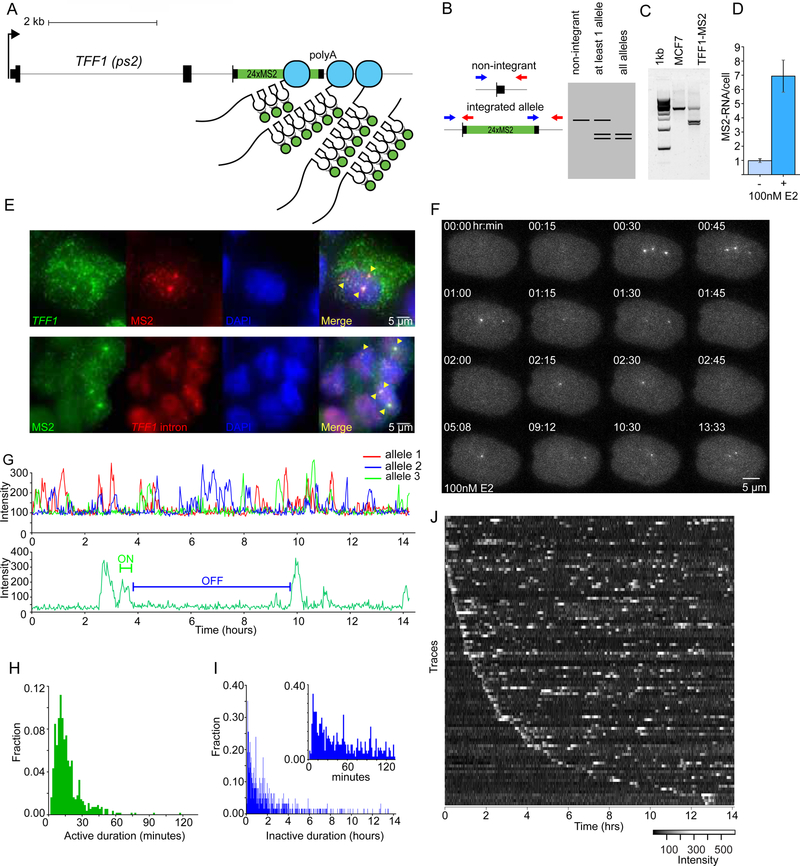
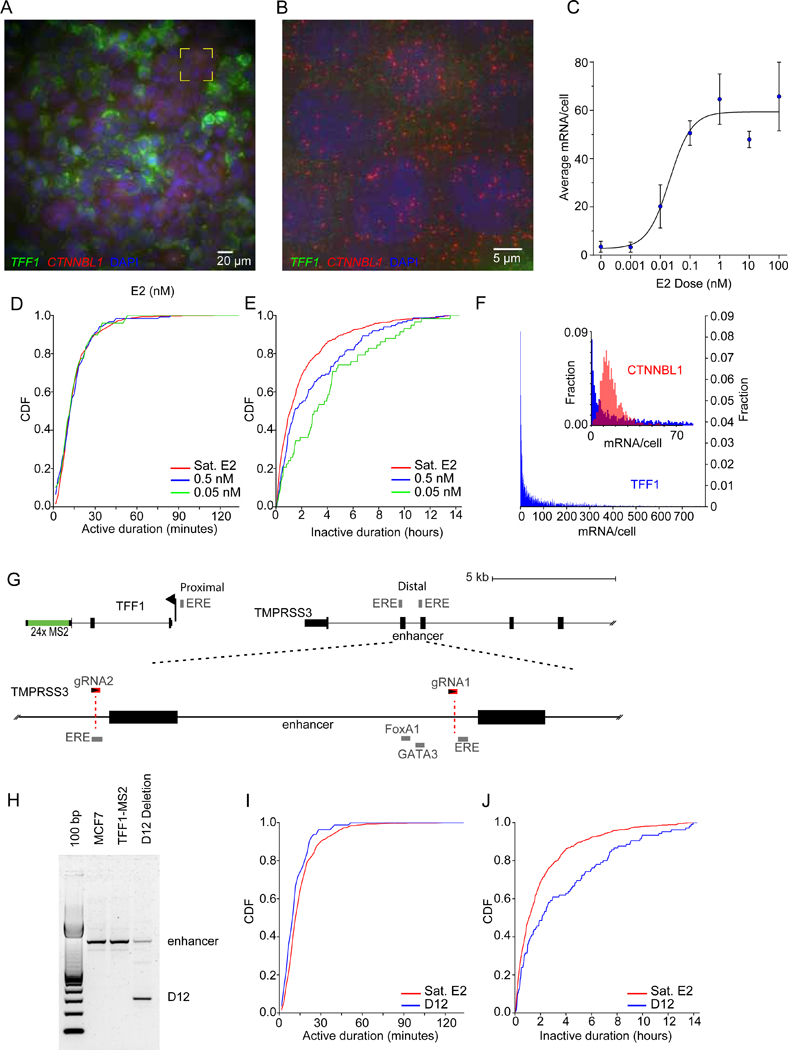
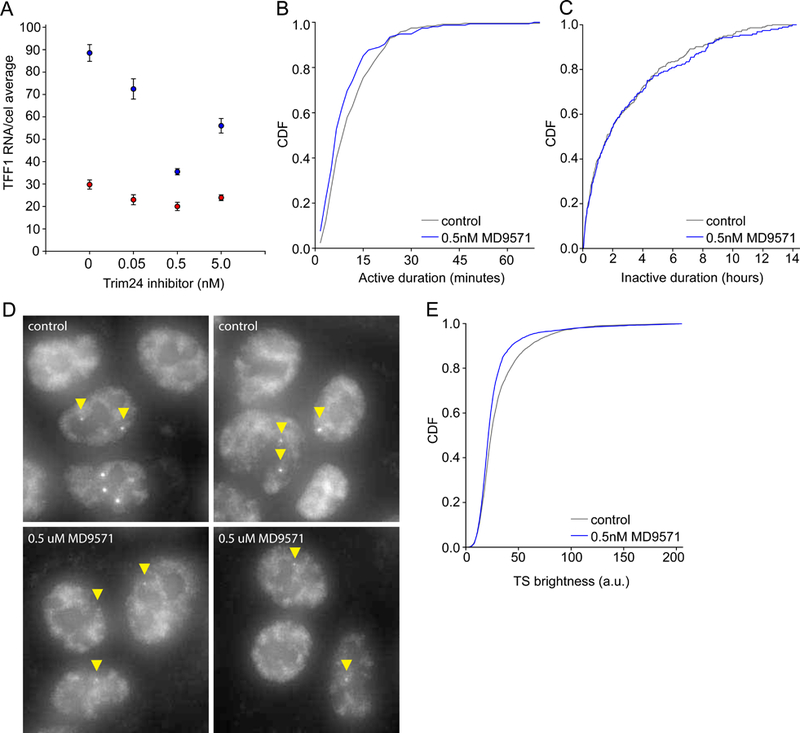
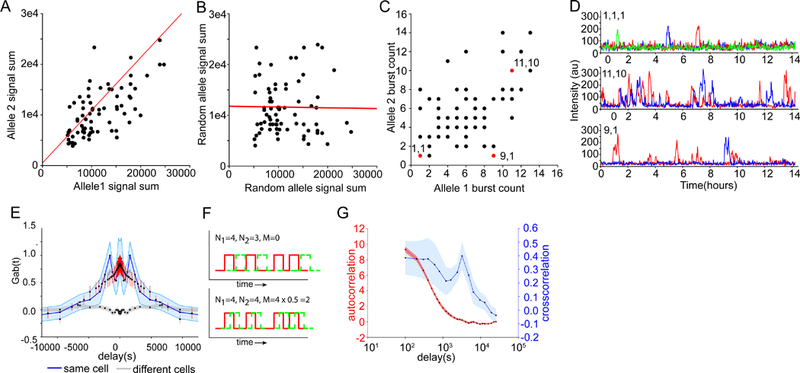
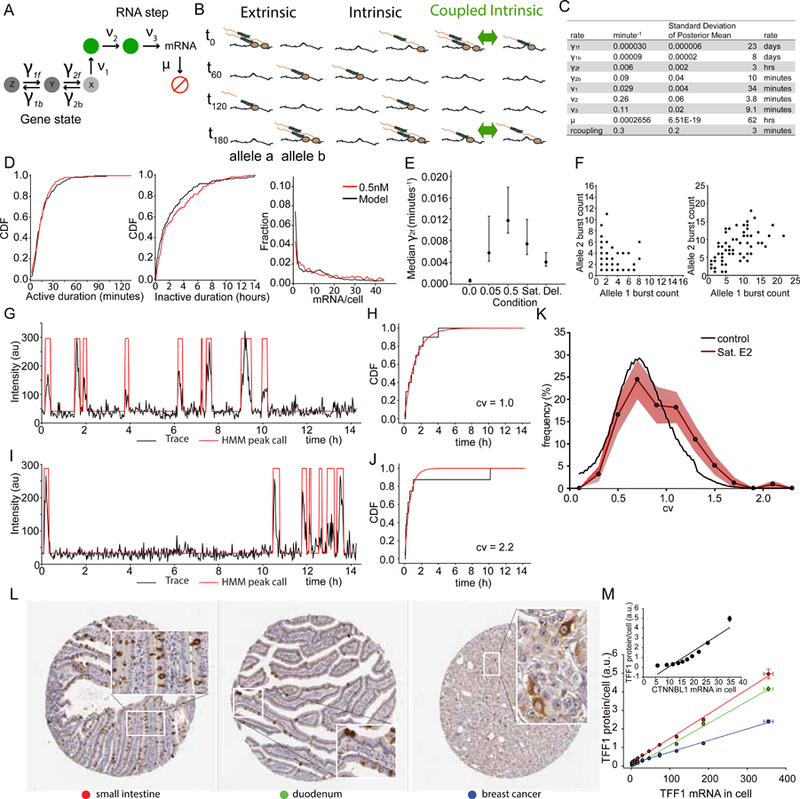
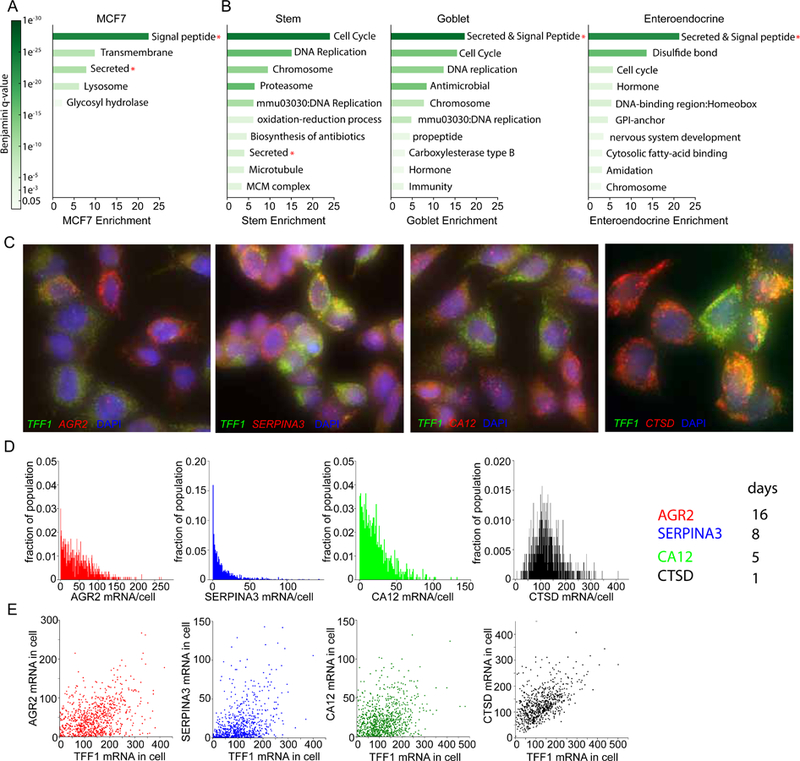
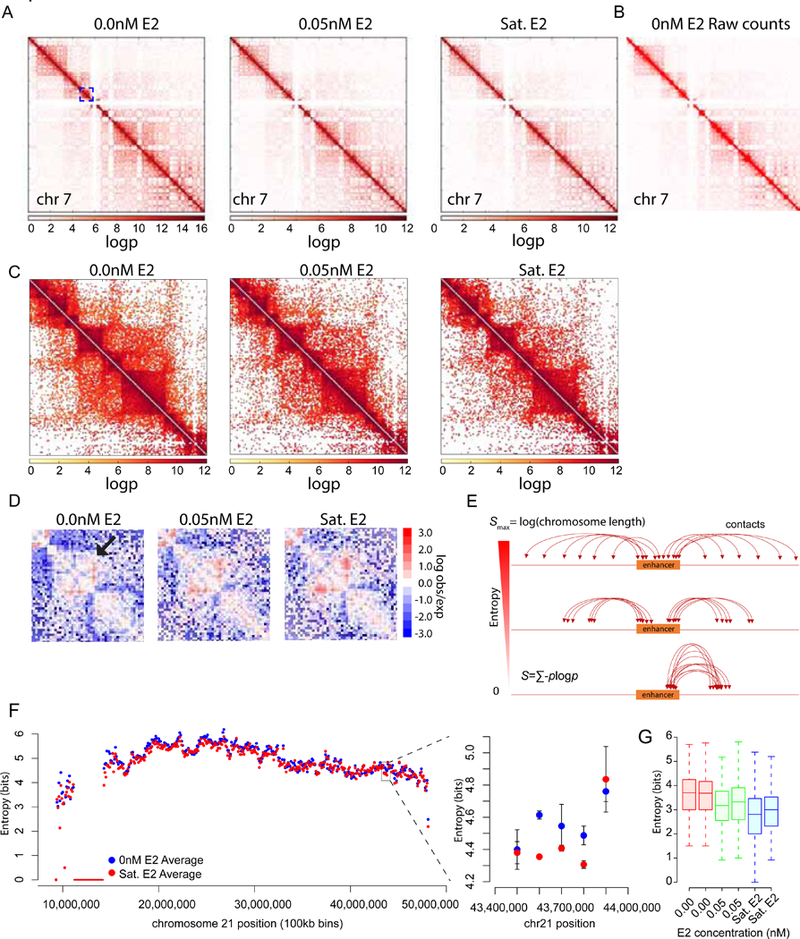
Transcriptional regulation in metazoans occurs through long-range genomic contacts between enhancers and promoters, and most genes are transcribed in episodic "bursts" of RNA synthesis. To understand the relationship between these two phenomena and the dynamic regulation of genes in response to upstream signals, we describe the use of live-cell RNA imaging coupled with Hi-C measurements and dissect the endogenous regulation of the estrogen-responsive TFF1 gene. Although TFF1 is highly induced, we observe short active periods and variable inactive periods ranging from minutes to days. The heterogeneity in inactive times gives rise to the widely observed "noise" in human gene expression and explains the distribution of protein levels in human tissue. We derive a mathematical model of regulation that relates transcription, chromosome structure, and the cell's ability to sense changes in estrogen and predicts that hypervariability is largely dynamic and does not reflect a stable biological state.
Keywords: RNA; chromosome; estrogen; fluorescence; heterogeneity; imaging; live-cell; single-molecule; steroid; transcription.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests
The authors declare no competing financial interests.
Figures
References
-
- Battich N, Stoeger T, and Pelkmans L (2015). Control of Transcript Variability in Single Mammalian Cells. Cell 163, 1596–1610. - PubMed
-
- Bezanson J, Edelman A, Karpinski S, and Shah VB (2017). Julia: A Fresh Approach to Numerical Computing. Siam Rev 59, 65–98.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
