

A Contraction Stress Model of Hypertrophic Cardiomyopathy due to Sarcomere Mutations
- PMID: 30554920
- PMCID: PMC6335568
- DOI: 10.1016/j.stemcr.2018.11.015
A Contraction Stress Model of Hypertrophic Cardiomyopathy due to Sarcomere Mutations
Abstract
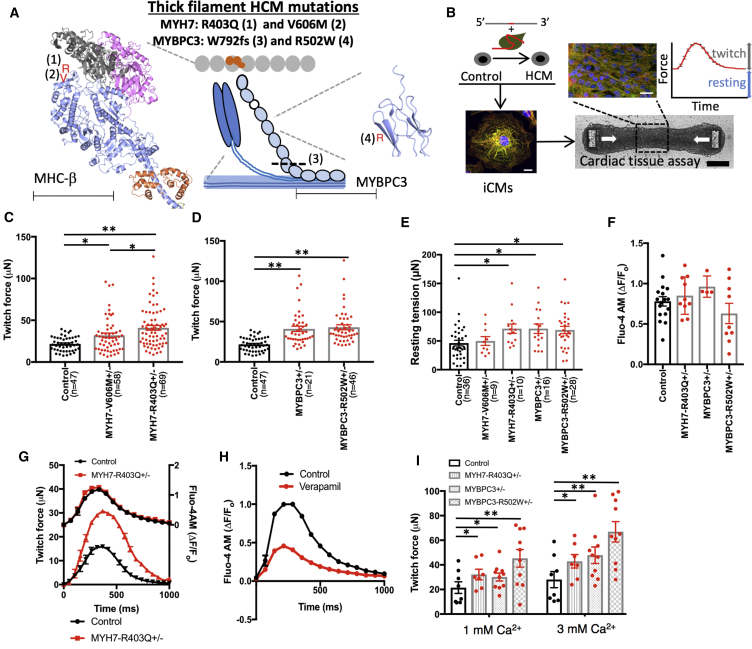
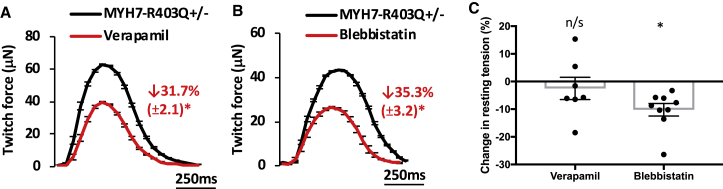
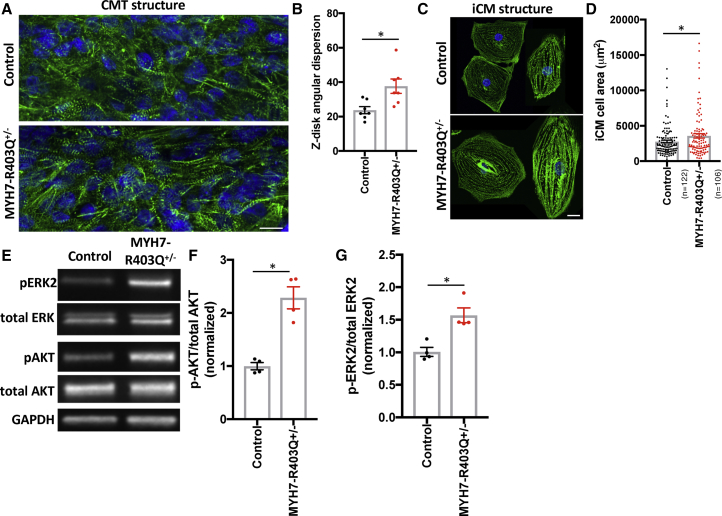
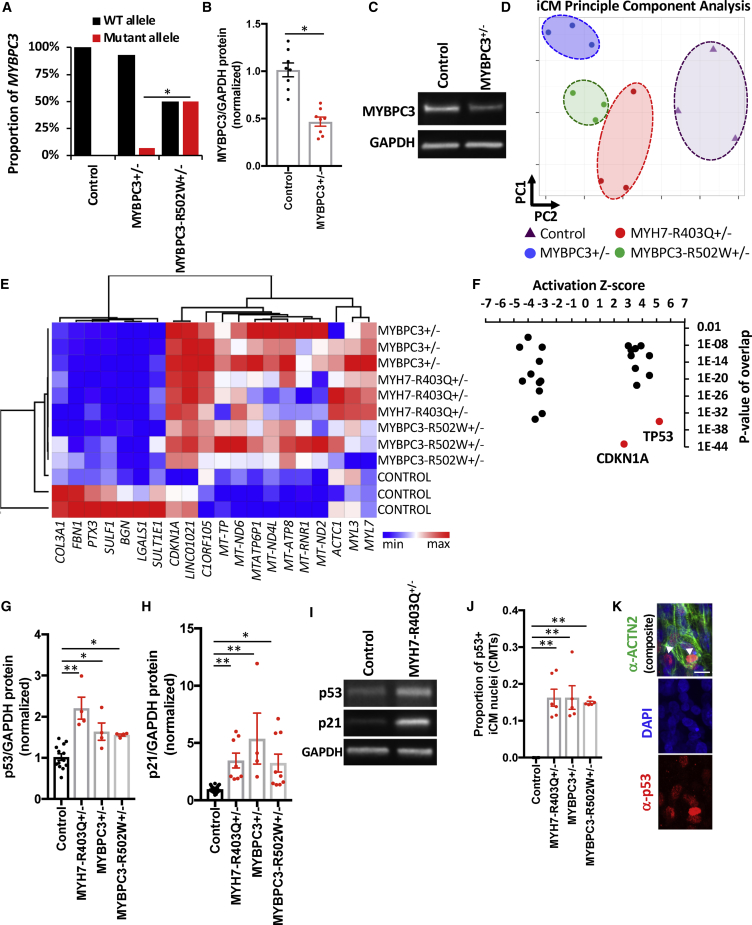
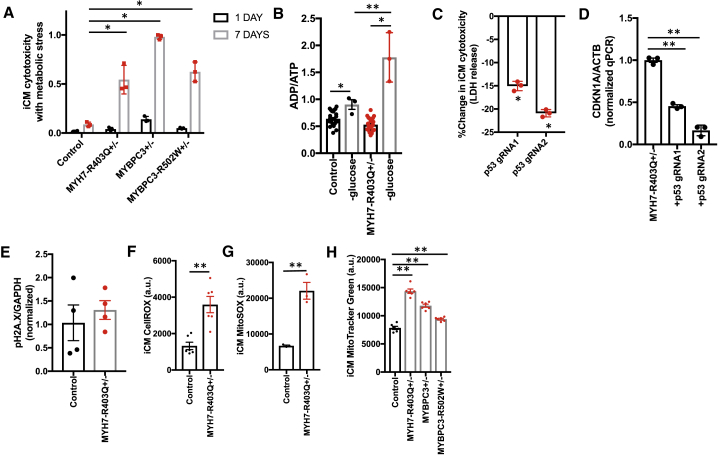
Thick-filament sarcomere mutations are a common cause of hypertrophic cardiomyopathy (HCM), a disorder of heart muscle thickening associated with sudden cardiac death and heart failure, with unclear mechanisms. We engineered four isogenic induced pluripotent stem cell (iPSC) models of β-myosin heavy chain and myosin-binding protein C3 mutations, and studied iPSC-derived cardiomyocytes in cardiac microtissue assays that resemble cardiac architecture and biomechanics. All HCM mutations resulted in hypercontractility with prolonged relaxation kinetics in proportion to mutation pathogenicity, but not changes in calcium handling. RNA sequencing and expression studies of HCM models identified p53 activation, oxidative stress, and cytotoxicity induced by metabolic stress that can be reversed by p53 genetic ablation. Our findings implicate hypercontractility as a direct consequence of thick-filament mutations, irrespective of mutation localization, and the p53 pathway as a molecular marker of contraction stress and candidate therapeutic target for HCM patients.
Keywords: cardiomyopathy; heart failure; hypertrophyp53 signaling; induced pluripotent stem cells; sarcomere function; tissue engineering.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Ashrafian H., McKenna W.J., Watkins H. Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ. Res. 2011;109:86–96. - PubMed
-
- Blankenburg R., Hackert K., Wurster S., Deenen R., Seidman J.G., Seidman C.E., Lohse M.J., Schmitt J.P. β-Myosin heavy chain variant Val606Met causes very mild hypertrophic cardiomyopathy in mice, but exacerbates HCM phenotypes in mice carrying other HCM mutations. Circ. Res. 2014;115:227–237. - PMC - PubMed
-
- Bulkley B.H., Weisfeldt M.L., Hutchins G.M. Asymmetric septal hypertrophy and myocardial fiber disarray. Features of normal, developing, and malformed hearts. Circulation. 1977;56:292–298. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
