

Clinical and molecular analysis of epilepsy-related genes in patients with Dravet syndrome
- PMID: 30558019
- PMCID: PMC6320057
- DOI: 10.1097/MD.0000000000013565
Clinical and molecular analysis of epilepsy-related genes in patients with Dravet syndrome
Abstract
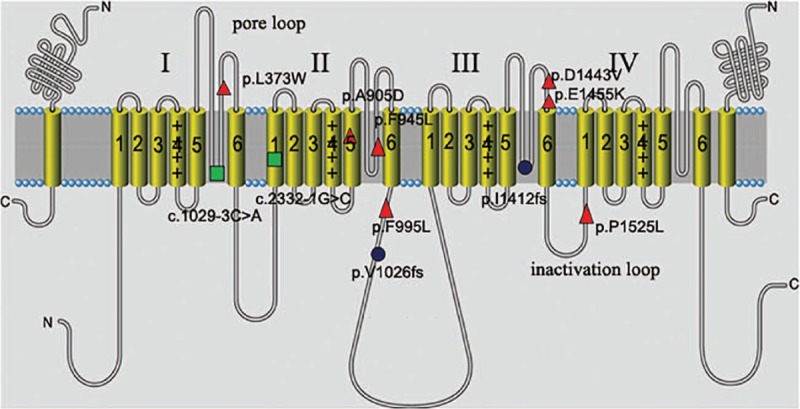
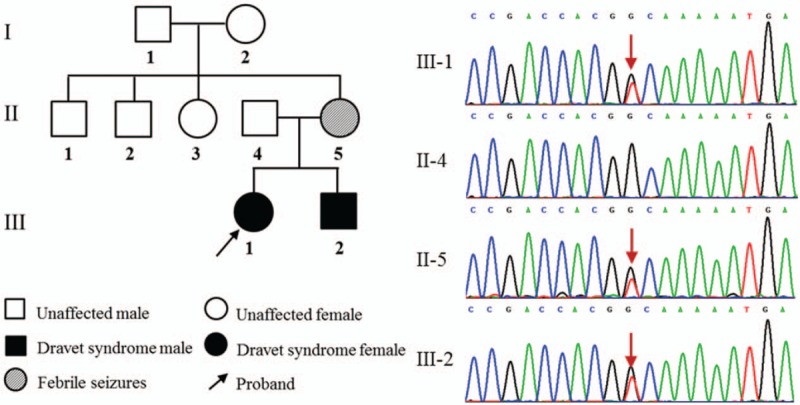
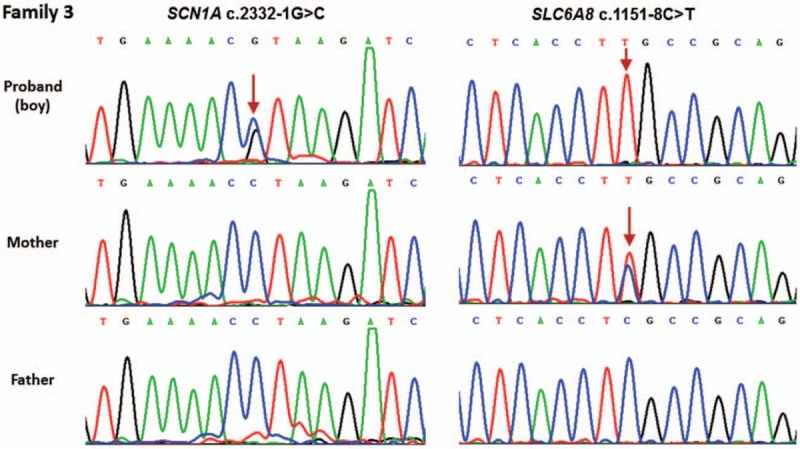
Dravet syndrome is considered to be one of the most severe types of genetic epilepsy. Mutations in SCN1A gene have been found to be responsible for at least 80% of patients with Dravet syndrome, and 90% of these mutations arise de novo. The variable clinical phenotype is commonly observed among these patients with SCN1A mutations, suggesting that genetic modifiers may influence the phenotypic expression of Dravet syndrome. In the present study, we described the clinical, pathological, and molecular characteristics of 13 Han Chinese pedigrees clinically diagnosed with Dravet syndrome. By targeted-exome sequencing, bioinformatics analysis and Sanger sequencing verification, 11 variants were identified in SCN1A gene among 11 pedigrees including 7 missense mutations, 2 splice site mutations, and 2 frameshift mutations (9 novel variants and 2 reported mutations). Particularly, 2 of these Dravet syndrome patients with SCN1A variants also harbored SCN9A, KCNQ2, or SLC6A8 variants. In addition, 2 subjects were failed to detect any pathogenic mutations in SCN1A and other epilepsy-related genes. These data suggested that SCN1A variants account for about 84.6% of Dravet syndrome in our cohort. This study expanded the mutational spectrum for the SCN1A gene, and also provided clinical and genetic evidence for the hypothesis that genetic modifiers may contribute to the variable manifestation of Dravet syndrome patients with SCN1A mutations. Thus, targeted-exome sequencing will make it possible to detect the interactions of epilepsy-related genes and reveal their modification on the severity of SCN1A mutation-related Dravet syndrome.
Conflict of interest statement
The authors have no conflicts of interest to disclose.
Figures
References
-
- Rosander C, Hallbook T. Dravet syndrome in Sweden: a population-based study. Dev Med Child Neurol 2015;57:628–33. - PubMed
-
- Bayat A, Hjalgrim H, Moller RS. The incidence of SCN1A-related Dravet syndrome in Denmark is 1: 22,000: a population-based study from 2004 to 2009. Epilepsia 2015;56:E36–9. - PubMed
-
- Meng H, Xu HQ, Yu L, et al. The SCN1A mutation database: updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Hum Mutat 2015;36:573–80. - PubMed
-
- Ishii A, Kanaumi T, Sohda M, et al. Association of nonsense mutation in GABRG2 with abnormal trafficking of GABA (A) receptors in severe epilepsy. Epilepsy Res 2014;108:420–32. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
