

A novel SLC12A3 homozygous c2039delG mutation in Gitelman syndrome with hypocalcemia
- PMID: 30558554
- PMCID: PMC6296056
- DOI: 10.1186/s12882-018-1163-3
A novel SLC12A3 homozygous c2039delG mutation in Gitelman syndrome with hypocalcemia
Abstract
Background: Gitelman syndrome (GS) is a rare autosomal recessive renal tubular disease, caused by mutations in the SLC12A3 gene, which encodes the renal thiazide-sensitive Na/Cl cotransporter (NCCT) in the distal renal tubule.
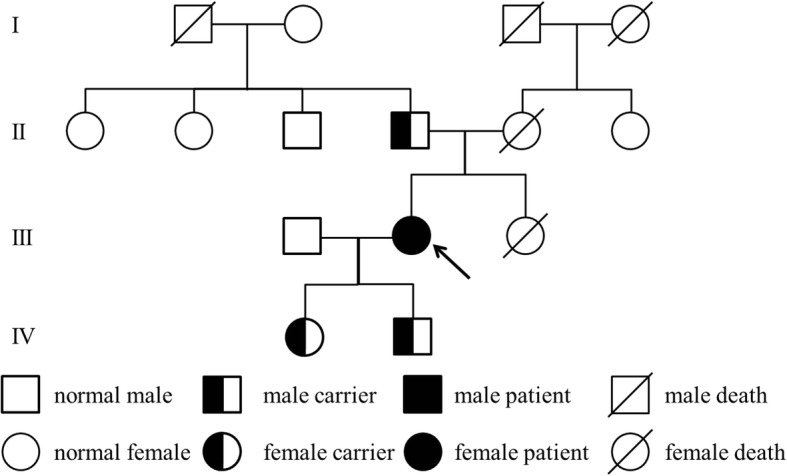
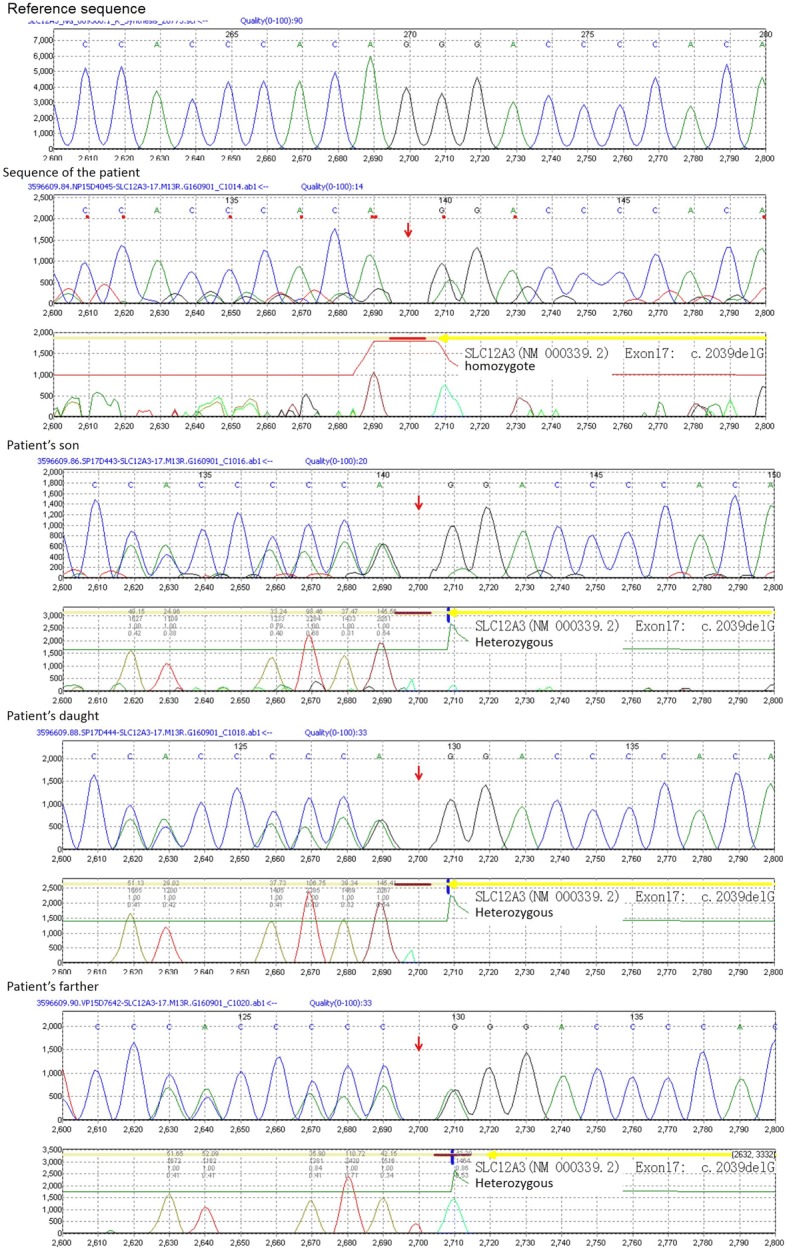
Case presentation: A 23-year-old woman was admitted with limb numbness, recurrent tetany and palpitation. Laboratory tests showed hypokalemic alkalosis, hypomagnesemia, hypocalcemia and secondary hyperaldosteronism, as well as hypocalciuria and transient decreased PTH. Next-generation sequencing detected a novel homozygous mutations c.2039delG in the SLC12A3 gene, and her father and children were all heterozygous carriers.
Conclusion: We reported a case of GS with a novel homozygous frame-shift mutation of SLC12A3, and reviewed recent literatures about diagnosis, differential diagnosis and treatments. Hypocalcemia in Gitelman syndrome is rare, and may be related to inhibited PTH secretion induced by hypomagnesemia.
Keywords: Gitelman syndrome; Hypocalcemia; SLC12A3 gene mutation.
Conflict of interest statement
Ethics approval and consent to participate
No ethical approval was required for this case report. Written informed consent was obtained from all individual participants or the guardians (if under age of 18) included in the study.
Consent for publication
Written informed consent was obtained from all individuals who participated to publish the details in this case.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Gitelman HJ, Graham JB. Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Phys. 1966;79:221–235. - PubMed
-
- Zhang L, Zhang J, Yang J, et al. PriVar: a toolkit for prioritizing SNVs and indels from next-generation sequencing data. Bioinformatics. 2013;29(1):124–125. - PubMed
-
- Parmar MS, Bhimji SS. Gitelman Syndrome. 2017.
-
- Blanchard A, Bockenhauer D, Bolignano D, et al. Gitelman syndrome: consensus and guidance from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. 2017;91(1):24–33. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
