

Reframing gene essentiality in terms of adaptive flexibility
- PMID: 30558585
- PMCID: PMC6296033
- DOI: 10.1186/s12918-018-0653-z
Reframing gene essentiality in terms of adaptive flexibility
Abstract
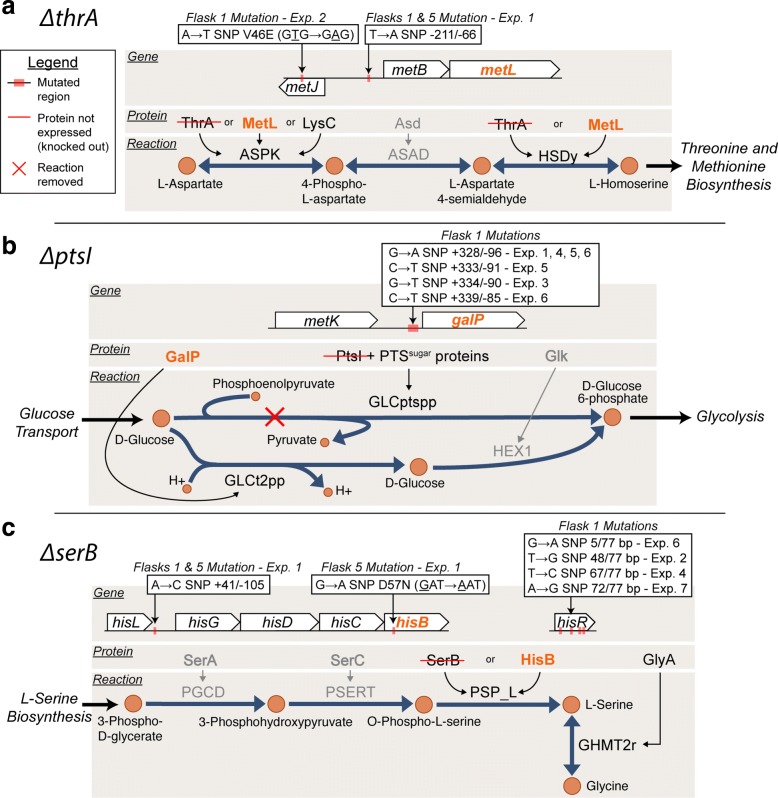
Background: Essentiality assays are important tools commonly utilized for the discovery of gene functions. Growth/no growth screens of single gene knockout strain collections are also often utilized to test the predictive power of genome-scale models. False positive predictions occur when computational analysis predicts a gene to be non-essential, however experimental screens deem the gene to be essential. One explanation for this inconsistency is that the model contains the wrong information, possibly an incorrectly annotated alternative pathway or isozyme reaction. Inconsistencies could also be attributed to experimental limitations, such as growth tests with arbitrary time cut-offs. The focus of this study was to resolve such inconsistencies to better understand isozyme activities and gene essentiality.
Results: In this study, we explored the definition of conditional essentiality from a phenotypic and genomic perspective. Gene-deletion strains associated with false positive predictions of gene essentiality on defined minimal medium for Escherichia coli were targeted for extended growth tests followed by population sequencing and transcriptome analysis. Of the twenty false positive strains available and confirmed from the Keio single gene knock-out collection, 11 strains were shown to grow with longer incubation periods making these actual true positives. These strains grew reproducibly with a diverse range of growth phenotypes. The lag phase observed for these strains ranged from less than one day to more than 7 days. It was found that 9 out of 11 of the false positive strains that grew acquired mutations in at least one replicate experiment and the types of mutations ranged from SNPs and small indels associated with regulatory or metabolic elements to large regions of genome duplication. Comparison of the detected adaptive mutations, modeling predictions of alternate pathways and isozymes, and transcriptome analysis of KO strains suggested agreement for the observed growth phenotype for 6 out of the 9 cases where mutations were observed.
Conclusions: Longer-term growth experiments followed by whole genome sequencing and transcriptome analysis can provide a better understanding of conditional gene essentiality and mechanisms of adaptation to such perturbations. Compensatory mutations are largely reproducible mechanisms and are in agreement with genome-scale modeling predictions to loss of function gene deletion events.
Keywords: Adaptive evolution; Essentiality; Genome-scale model.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures



Similar articles
-
Model-driven discovery of underground metabolic functions in Escherichia coli.Proc Natl Acad Sci U S A. 2015 Jan 20;112(3):929-34. doi: 10.1073/pnas.1414218112. Epub 2015 Jan 6. Proc Natl Acad Sci U S A. 2015. PMID: 25564669 Free PMC article.
-
Potential confounding mutations in Keio knockout strains: implications for research accuracy.Microbiol Spectr. 2025 Mar 31;13(5):e0203624. doi: 10.1128/spectrum.02036-24. Online ahead of print. Microbiol Spectr. 2025. PMID: 40162750 Free PMC article.
-
High-throughput transposon mutagenesis in the family Enterobacteriaceae reveals core essential genes and rapid turnover of essentiality.mBio. 2024 Oct 16;15(10):e0179824. doi: 10.1128/mbio.01798-24. Epub 2024 Aug 29. mBio. 2024. PMID: 39207104 Free PMC article.
-
The conditional nature of gene essentiality.Curr Opin Genet Dev. 2019 Oct;58-59:55-61. doi: 10.1016/j.gde.2019.07.015. Epub 2019 Aug 27. Curr Opin Genet Dev. 2019. PMID: 31470233 Review.
-
Machine learning approach to gene essentiality prediction: a review.Brief Bioinform. 2021 Sep 2;22(5):bbab128. doi: 10.1093/bib/bbab128. Brief Bioinform. 2021. PMID: 33842944 Review.
Cited by
-
The Escherichia coli transcriptome mostly consists of independently regulated modules.Nat Commun. 2019 Dec 4;10(1):5536. doi: 10.1038/s41467-019-13483-w. Nat Commun. 2019. PMID: 31797920 Free PMC article.
-
Gene Dispensability in Escherichia coli Grown in Thirty Different Carbon Environments.mBio. 2020 Sep 29;11(5):e02259-20. doi: 10.1128/mBio.02259-20. mBio. 2020. PMID: 32994326 Free PMC article.
-
Path to improving the life cycle and quality of genome-scale models of metabolism.Cell Syst. 2021 Sep 22;12(9):842-859. doi: 10.1016/j.cels.2021.06.005. Cell Syst. 2021. PMID: 34555324 Free PMC article. Review.
-
The hallmarks of a tradeoff in transcriptomes that balances stress and growth functions.Res Sq [Preprint]. 2023 Apr 12:rs.3.rs-2729651. doi: 10.21203/rs.3.rs-2729651/v1. Res Sq. 2023. Update in: mSystems. 2024 Jul 23;9(7):e0030524. doi: 10.1128/msystems.00305-24. PMID: 37090546 Free PMC article. Updated. Preprint.
-
Exploring the Glucose Fluxotype of the E. coli y-ome Using High-Resolution Fluxomics.Metabolites. 2021 Apr 26;11(5):271. doi: 10.3390/metabo11050271. Metabolites. 2021. PMID: 33926117 Free PMC article.
References
-
- Mobegi FM, Zomer A, de Jonge MI, van Hijum SAFT. Advances and perspectives in computational prediction of microbial gene essentiality. Brief Funct Genomics. 2017;16(2):70–9. - PubMed
-
- Hutchison CA3rd, Chuang R-Y, Noskov VN, Assad-Garcia N, Deerinck TJ, Ellisman MH, Gill J, Kannan K, Karas BJ, Ma L, Pelletier JF, Qi Z-Q, Richter RA, Strychalski EA, Sun L, Suzuki Y, Tsvetanova B, Wise KS, Smith HO, Glass JI, Merryman C, Gibson DG, Venter JC. Design and synthesis of a minimal bacterial genome. Science. 2016;351(6280):6253. - PubMed
-
- Chung BK-S, Dick T, Lee D-Y. In silico analyses for the discovery of tuberculosis drug targets. J Antimicrob Chemother. 2013;68(12):2701–9. - PubMed
-
- Hughes D, Andersson DI. Evolutionary trajectories to antibiotic resistance. Annu Rev Microbiol. 2017;71:579–96. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
