

Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data
- PMID: 30558668
- PMCID: PMC6298009
- DOI: 10.1186/s40168-018-0605-2
Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data
Abstract
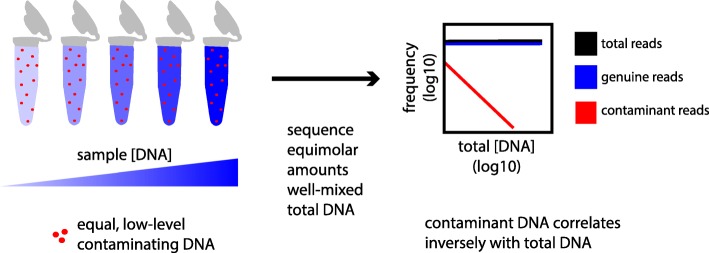
Background: The accuracy of microbial community surveys based on marker-gene and metagenomic sequencing (MGS) suffers from the presence of contaminants-DNA sequences not truly present in the sample. Contaminants come from various sources, including reagents. Appropriate laboratory practices can reduce contamination, but do not eliminate it. Here we introduce decontam ( https://github.com/benjjneb/decontam ), an open-source R package that implements a statistical classification procedure that identifies contaminants in MGS data based on two widely reproduced patterns: contaminants appear at higher frequencies in low-concentration samples and are often found in negative controls.
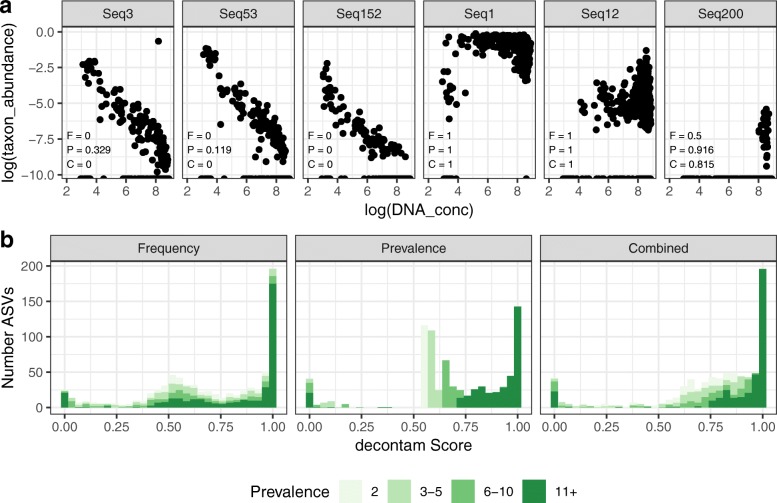
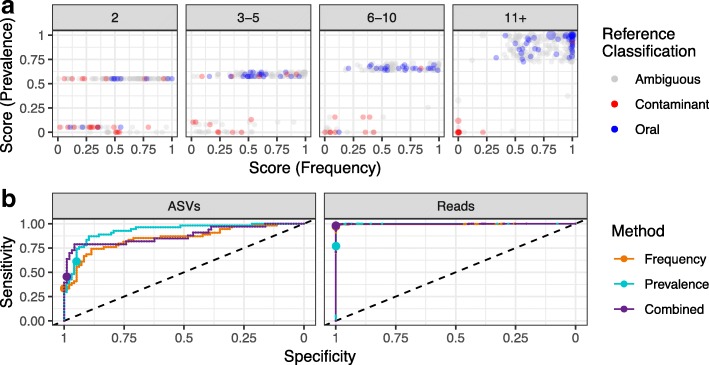
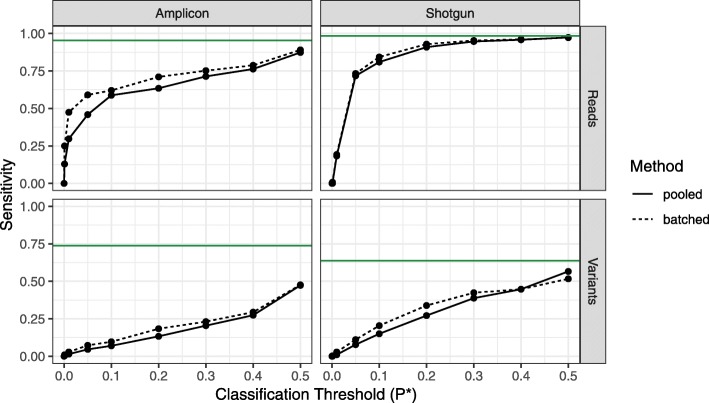
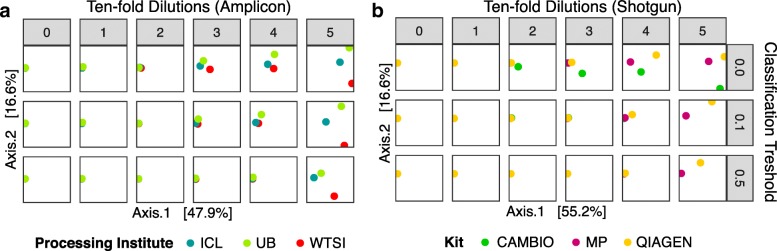
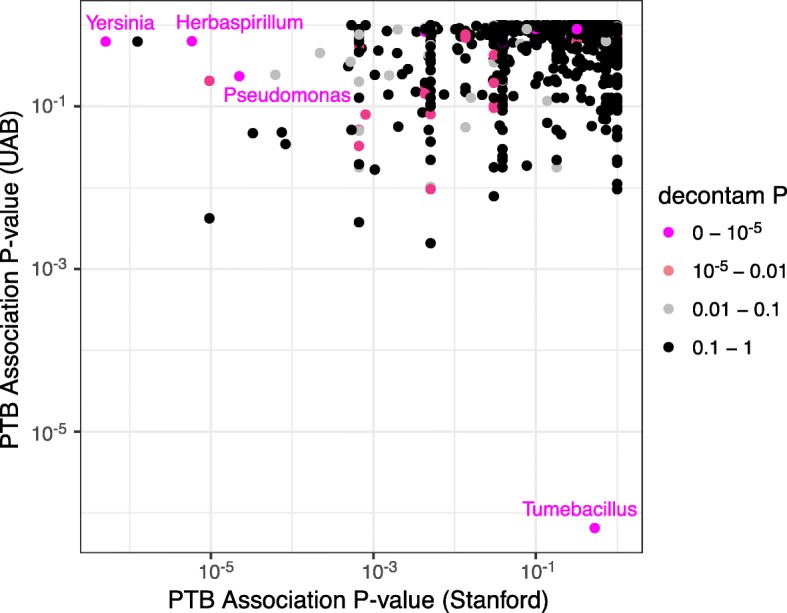
Results: Decontam classified amplicon sequence variants (ASVs) in a human oral dataset consistently with prior microscopic observations of the microbial taxa inhabiting that environment and previous reports of contaminant taxa. In metagenomics and marker-gene measurements of a dilution series, decontam substantially reduced technical variation arising from different sequencing protocols. The application of decontam to two recently published datasets corroborated and extended their conclusions that little evidence existed for an indigenous placenta microbiome and that some low-frequency taxa seemingly associated with preterm birth were contaminants.
Conclusions: Decontam improves the quality of metagenomic and marker-gene sequencing by identifying and removing contaminant DNA sequences. Decontam integrates easily with existing MGS workflows and allows researchers to generate more accurate profiles of microbial communities at little to no additional cost.
Keywords: 16S rRNA gene; DNA contamination; Marker-gene; Metagenomics; Microbiome.
Conflict of interest statement
Ethics approval and consent to participate
The study was approved by the Administrative Panels on Human Subjects Research (Stanford IRB protocol #21586) and by the Human Research Protection Program at UCSF (UCSF IRB protocol ##11-06283). All research subjects provided written informed consent prior to specimen collection.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Comment in
-
Towards precision quantification of contamination in metagenomic sequencing experiments.Microbiome. 2019 Apr 16;7(1):62. doi: 10.1186/s40168-019-0678-6. Microbiome. 2019. PMID: 30992055 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
