

Hot topics in Fabry disease
- PMID: 30559317
- PMCID: PMC6581083
- DOI: 10.1136/postgradmedj-2018-136056
Hot topics in Fabry disease
Abstract
Fabry disease is a rare inborn error of the enzyme α-galactosidase (α-Gal) and results in lysosomal substrate accumulation in tissues with a wide range of clinical presentations. The disease has attracted a lot of interest over the last years, in particular since enzyme replacement therapy (ERT) has become widely available in 2001. With rising awareness and rising numbers of (diagnosed) patients, physicians encounter new challenges. Over 900 α-Gal gene mutations are currently known, some with doubtful clinical significance, posing diagnostic and prognostic difficulties for the clinician and a lot of uncertainty for patients. Another challenge are patients who develop neutralising antibodies to ERT, which possibly leads to reduced therapy effectiveness. In this article, we summarise the latest developments in the science community regarding diagnostics and management of this rare lysosomal storage disorder and offer an outlook to future treatments.
Keywords: Fabry disease; antibodies; biomarkers; multidisciplinary working.
© Author(s) (or their employer(s)) 2018. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: TC has received travel assistance from Genzyme/Sanofi. JM has received travel assistance and speaker honoraria from Genzyme/Sanofi and travel assistance from Amicus Therapeutics and Shire. JG and LS have nothing to declare. PN has received honoraria for lecturing and advisory board participation from Amicus Therapeutics, Genzyme/Sanofi and Shire. CW has received honoraria for lecturing and advisory board participation from Genzyme/Sanofi, Actelion and Protalix and Shire. Research grants were given to the Institution from Genzyme/Sanofi and Shire.
Figures
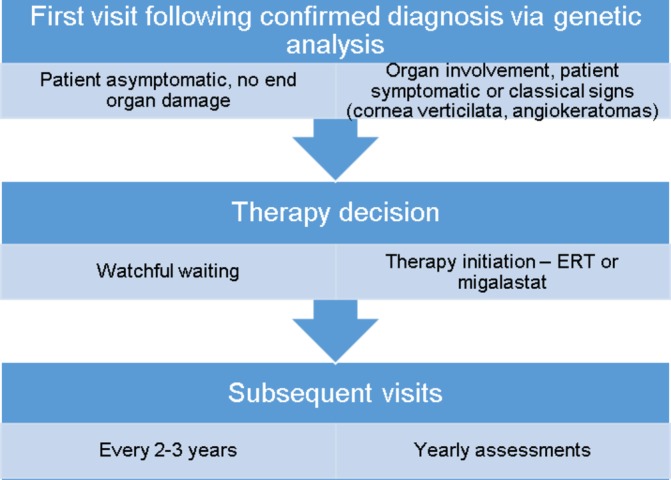
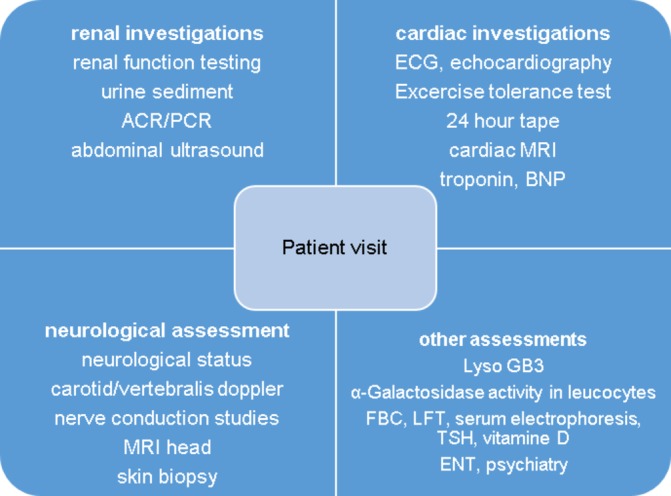
References
-
- Desnick RJ, Astrin KH, Bishop DF. Fabry disease: molecular genetics of the inherited nephropathy. Adv Nephrol Necker Hosp 1989;18:113–27. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
