

Hyperactivation of Oncogenic JAK3 Mutants Depend on ATP Binding to the Pseudokinase Domain
- PMID: 30560087
- PMCID: PMC6287396
- DOI: 10.3389/fonc.2018.00560
Hyperactivation of Oncogenic JAK3 Mutants Depend on ATP Binding to the Pseudokinase Domain
Abstract
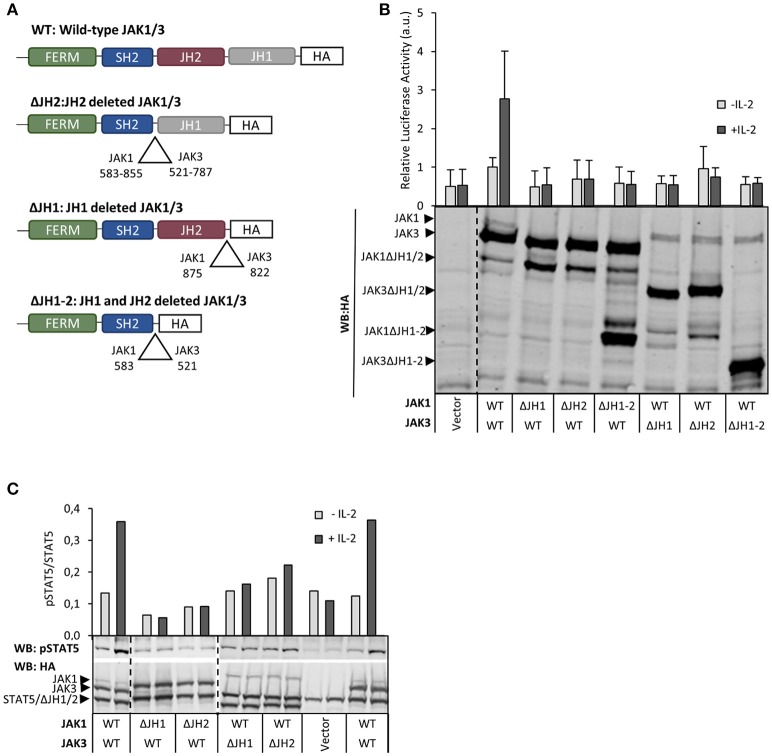
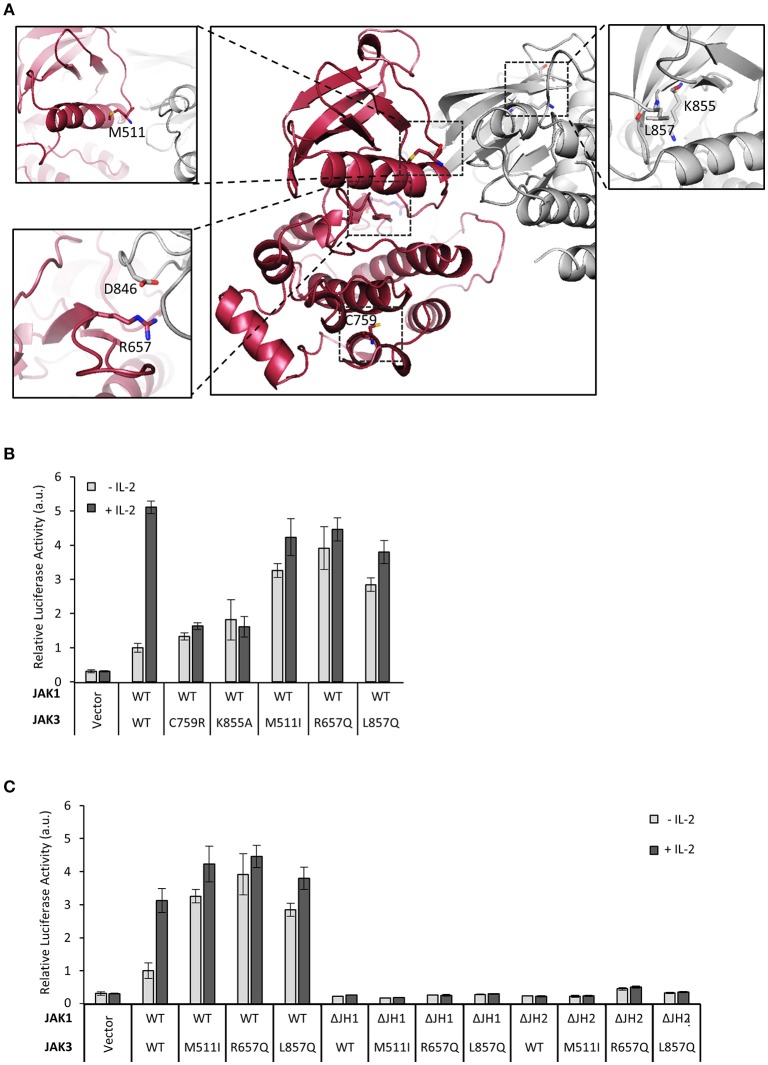
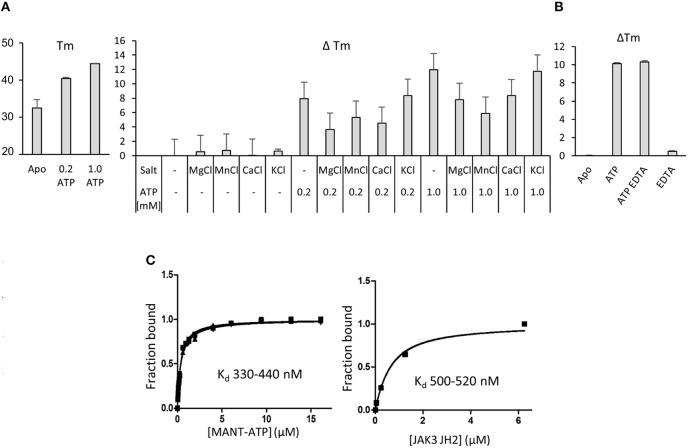
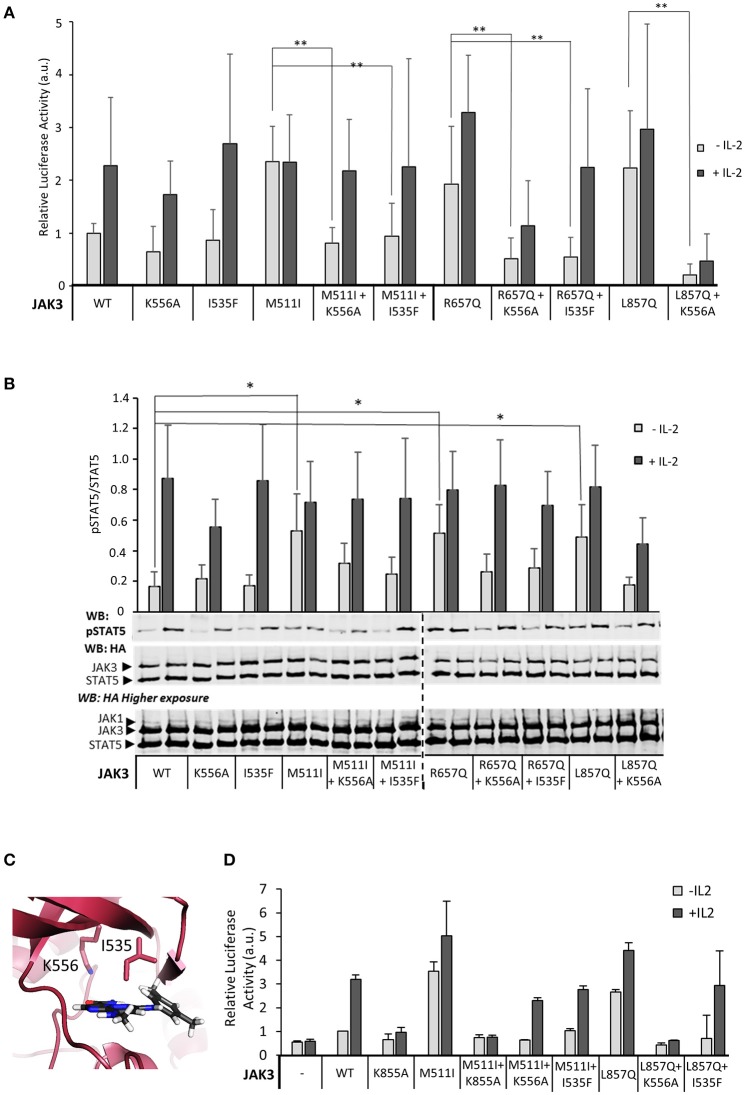
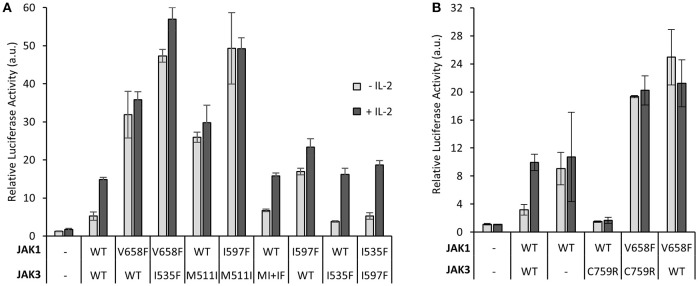
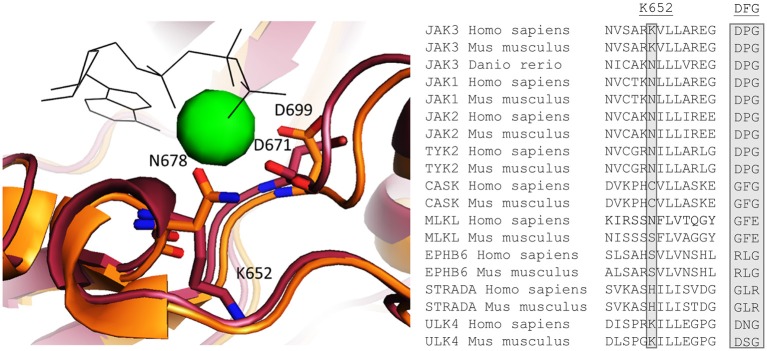
Janus kinase 3 (JAK3) tyrosine kinase has a central role in the control of lymphopoiesis, and mutations in JAK3 can lead to either severe combined immunodeficiency or leukemia and lymphomas. JAK3 associates with the common gamma chain (γc) receptor and functions in a heteromeric signaling pair with JAK1. In IL-2 signaling JAK1 is the effector kinase for STAT5 phosphorylation but the precise molecular regulatory mechanisms of JAK1 and JAK3 and their individual domains are not known. The pseudokinase domain (JAK homology 2, JH2) of JAK3 is of particular interest as approximately half of clinical JAK3 mutations cluster into it. In this study, we investigated the role of JH2s of JAK1 and JAK3 in IL-2R signaling and show that STAT5 activation requires both JH1 and JH2 of JAK1, while both JH1 and JH2 in JAK3 are specifically required for the cytokine-induction of cellular signaling. Characterization of recombinant JAK3 JH2 in thermal shift assay shows an unstable protein domain, which is strongly stabilized by ATP binding. Unexpectedly, nucleotide binding to JAK3 JH2 was found to be cation-independent. JAK3 JH2 showed higher nucleotide binding affinity in MANT-ATP and fluorescent polarization competition assays compared to the other JAK JH2s. Analysis of the functional role of ATP binding in JAK3 JH2 in cells and in zebrafish showed that disruption of ATP binding suppresses ligand-independent activation of clinical JAK3 gain-of-function mutations residing in either JH2 or JH1 but does not inhibit constitutive activation of oncogenic JAK1. ATP-binding site mutations in JAK3 JH2 do not, however, abrogate normal IL-2 signaling making them distinct from JH2 deletion or kinase-deficient JAK3. These findings underline the importance of JAK3 JH2 for cellular signaling in both ligand-dependent and in gain-of-function mutation-induced activation. Furthermore, they identify the JH2 ATP-binding site as a key regulatory region for oncogenic JAK3 signaling, and thus a potential target for therapeutic modulation.
Keywords: JAK kinase; cytokine; leukemia; nucleotide binding; pseudokinase.
Figures
References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous