

Vasculitis in Systemic Autoinflammatory Diseases
- PMID: 30560109
- PMCID: PMC6287042
- DOI: 10.3389/fped.2018.00377
Vasculitis in Systemic Autoinflammatory Diseases
Abstract
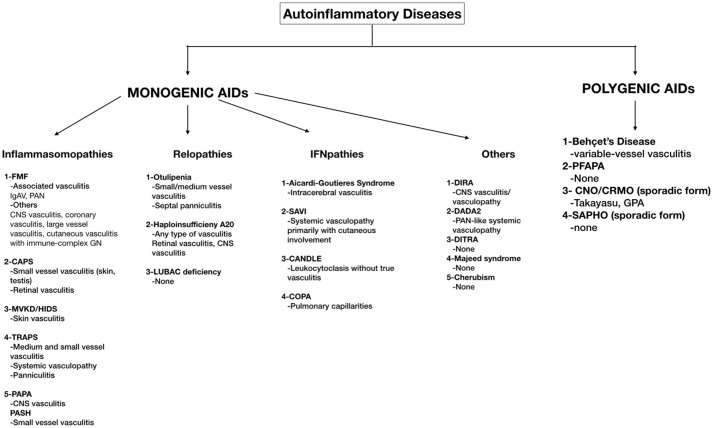
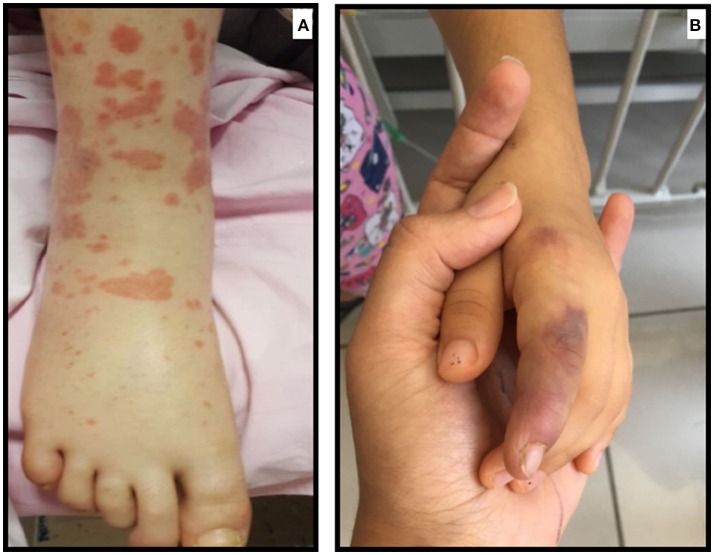
Autoinflammatory diseases (AID) are diseases of the innate immune system, characterized by recurrent episodes of localized or systemic inflammation. Vasculitis may accompany AID. The causes of the association of vasculitis with monogenic AID are still debated. Among the monogenic AID, Familial Mediterranean Fever (FMF) is the most common. IgA-related vasculitis (IgAV) and Polyarteritis Nodosa (PAN) involving small and/or medium-sized vessels have an increased frequency among FMF patients. There are also case reports revealing vasculitic features in Cryopyrin-Associated Periodic Fever Syndrome (CAPS), Tumor Necrosis Factor Receptor-Associated Periodic Syndrome (TRAPS), Mevalonate Kinase Deficiency (MKD), also known as Hyper IgD syndrome (HIDS), Deficiency of IL-1 Receptor Antagonist (DIRA) and Pyogenic Arthritis, Pyoderma gangrenosum, and Acne (PAPA) patients. Central nervous system vasculitis and vasculopathy have been reported in DIRA and PAPA patients whereas small vessel involvement affecting skin has been reported in CAPS, TRAPS, and MKD patients. Alternatively, vasculitis can also be a leading feature especially in the recently defined monogenic AID (Otulipenia, Deficiency of Adenosine Deaminase 2-DADA2, Haploinsufficiency of A20) and interferonopathies (STING-associated vasculopathy with onset in infancy-SAVI). DADA2 often presents as a PAN-like disease. In otulipenia, patients have painful subcutaneous nodules caused by septal panniculitis with small and medium vessel vasculitis. Haploinsufficiency of A20 (also called Familial Behcet-like Autoinflammatory Syndrome) results in a phenotype very similar to the variable vessel vasculitis of Behcet's disease with recurrent oral-genital ulcers, in addition to, skin rash, uveitis, and polyarthritis. SAVI is an autoinflammatory vasculopathy with increased Interferon (IFN) signature, causing severe skin lesions resulting in ulceration, necrosis, and in some cases, amputation. Behcet's Disease (BD) is a multifactorial polygenic AID characterized by recurrent attacks of oral-genital ulcers, skin lesions, uveitis and a unique vasculitis affecting both arteries and veins of all sizes. Many clinical features overlap with other autoinflammatory diseases and overexpression of proinflammatory cytokines is an important feature of the disease.
Keywords: Behcet disease; autoinflammatory diseases; inflammasomopathies; interferonopathies; relopathies; vasculitis.
Figures
References
-
- Ehrlich P. Studies in Immunity. London: Wiley; (1910).
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous