

SCAPER-associated nonsyndromic autosomal recessive retinitis pigmentosa
- PMID: 30561111
- PMCID: PMC6349500
- DOI: 10.1002/ajmg.a.61001
SCAPER-associated nonsyndromic autosomal recessive retinitis pigmentosa
Abstract
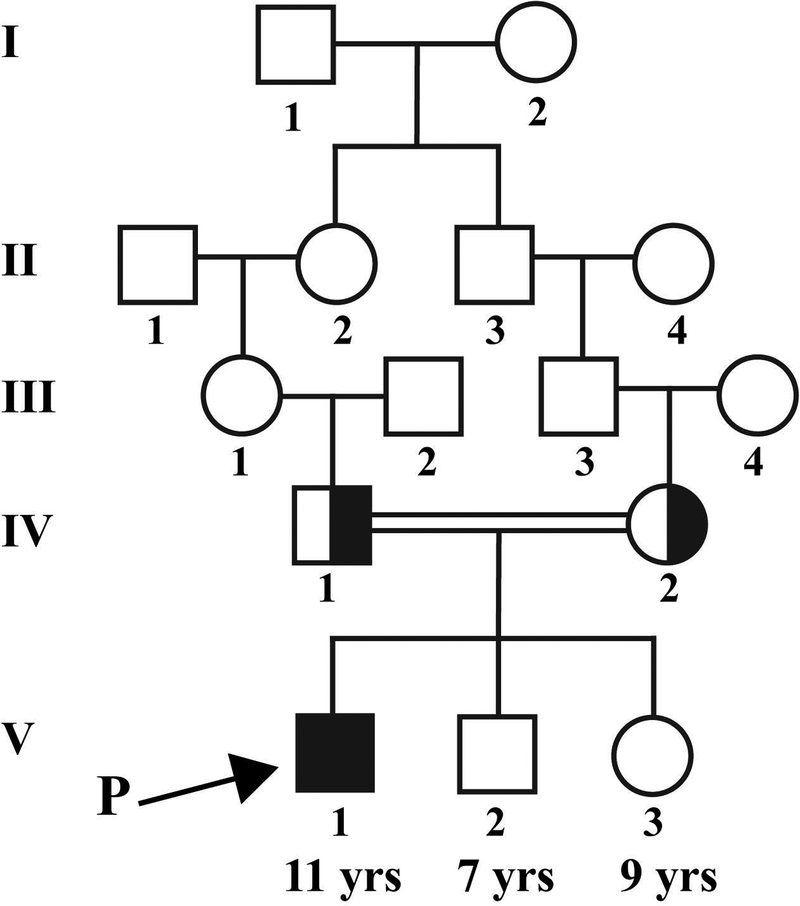
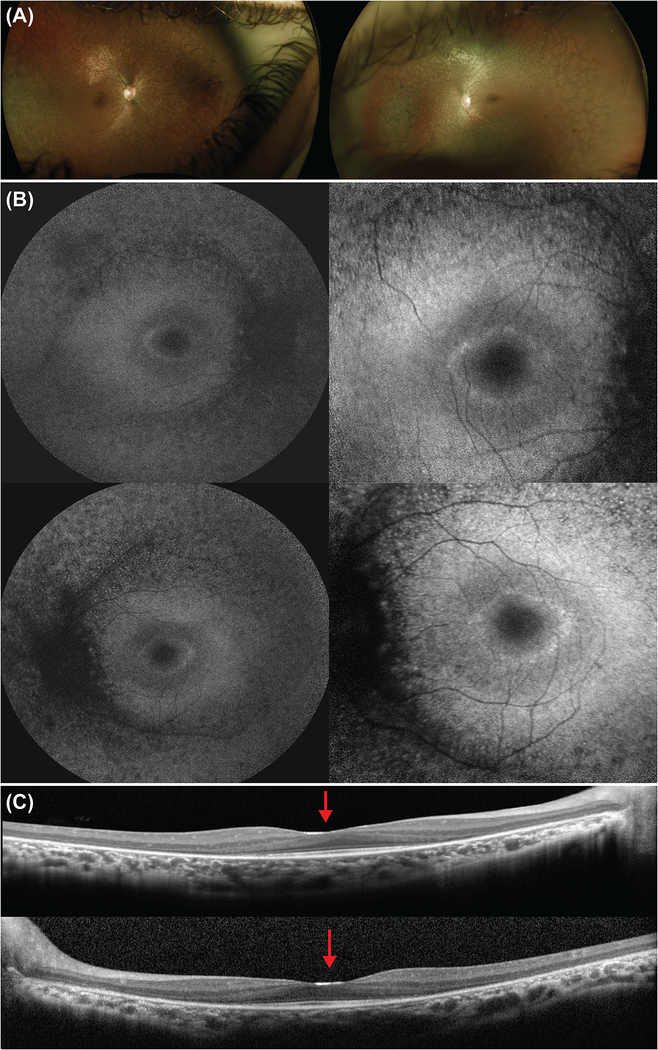
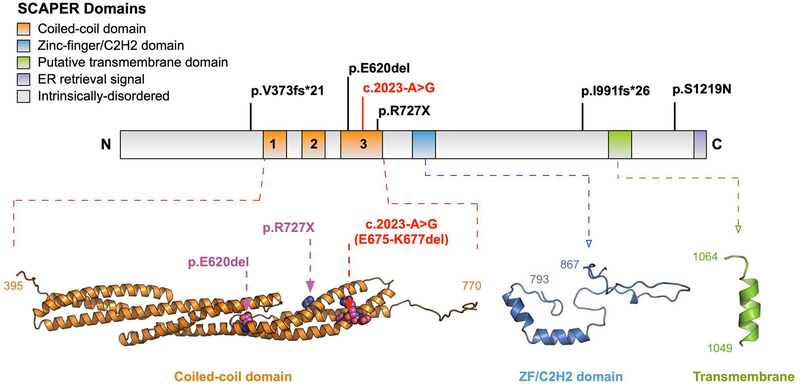
Mutations in the gene SCAPER (S-phase CyclinA Associated Protein residing in the Endoplasmic Reticulum) have recently been identified as causing syndromic autosomal recessive retinitis pigmentosa with the extraocular manifestations of intellectual disability and attention-deficit/hyperactivity disorder. We present the case of an 11-year-old boy that presented to our clinic with the complaint of decreased night vision. Clinical presentation, family history, and diagnostic imaging were congruent with the diagnosis of autosomal recessive retinitis pigmentosa. Genetic testing of the patient and both parents via whole-exome sequencing revealed the homozygous mutation c.2023-2A>G in SCAPER. Unique to our patient's presentation is the absence of intellectual disability and attention-deficit/hyperactivity disorder, suggesting that SCAPER-associated retinitis pigmentosa can also present without systemic manifestations.
Keywords: SCAPER; autosomal recessive; retinitis pigmentosa; syndromic disorder.
© 2018 Wiley Periodicals, Inc.
Conflict of interest statement
Figures
References
-
- DeLano WL (2002). The PyMOL molecular graphics system. http://www.pymol.org/. Retrieved from http://www.pymol.org/
-
- Estrada-Cuzcano A, Koenekoop RK, Senechal A, De Baere EB, de Ravel T, Banfi S, … Klevering BJ (2012). BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet-Biedl syndrome. Arch Ophthalmol, 130(11), 1425–1432. doi:10.1001/archophthalmol.2012.2434 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R21 AG050437/AG/NIA NIH HHS/United States
- R01 EY018213/EY/NEI NIH HHS/United States
- Medical Student Eye Research FellowshipPhysician-Scientist Award/Research to Prevent Blindness/International
- P30 EY026877/EY/NEI NIH HHS/United States
- R01 EY024665/EY/NEI NIH HHS/United States
- Research to Prevent Blindness/International
- K08 EY020530/EY/NEI NIH HHS/United States
- R01 EY026682/EY/NEI NIH HHS/United States
- R01 EY024698/EY/NEI NIH HHS/United States
- P30 EY019007/EY/NEI NIH HHS/United States
- 2013103/DDCF/Doris Duke Charitable Foundation/United States
- R01 EY025225/EY/NEI NIH HHS/United States
- 5P30CA013696/CA/NCI NIH HHS/United States
- P30 CA013696/CA/NCI NIH HHS/United States
- T32 GM007337/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
