

Variable Protease-Sensitive Prionopathy Transmission to Bank Voles
- PMID: 30561322
- PMCID: PMC6302590
- DOI: 10.3201/eid2501.180807
Variable Protease-Sensitive Prionopathy Transmission to Bank Voles
Abstract
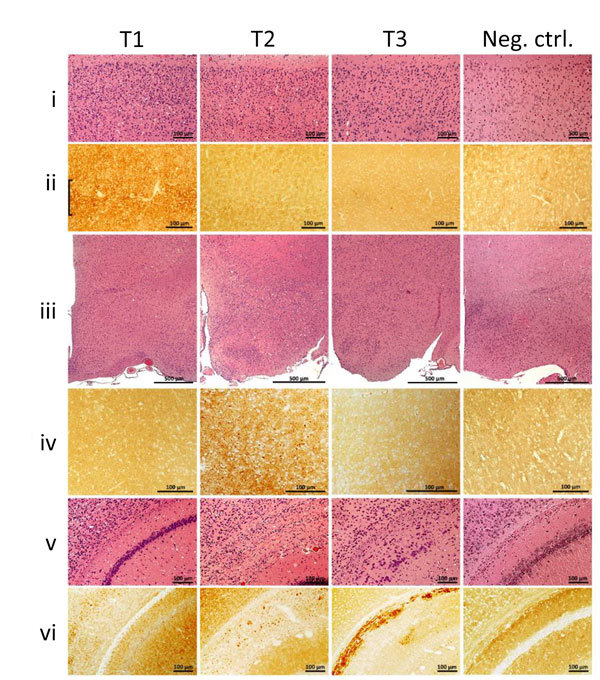
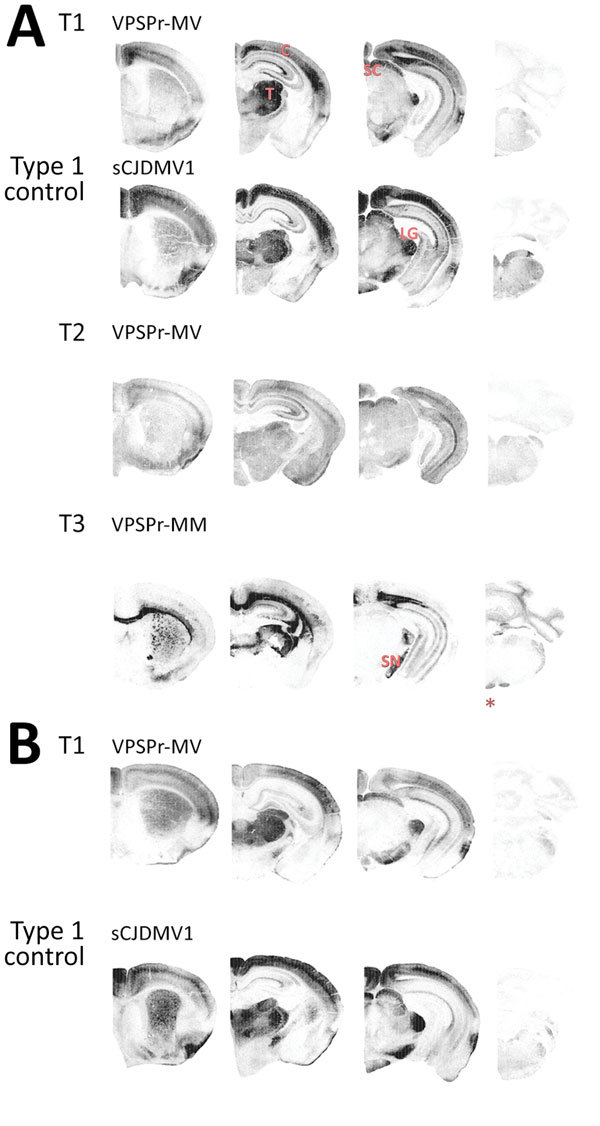
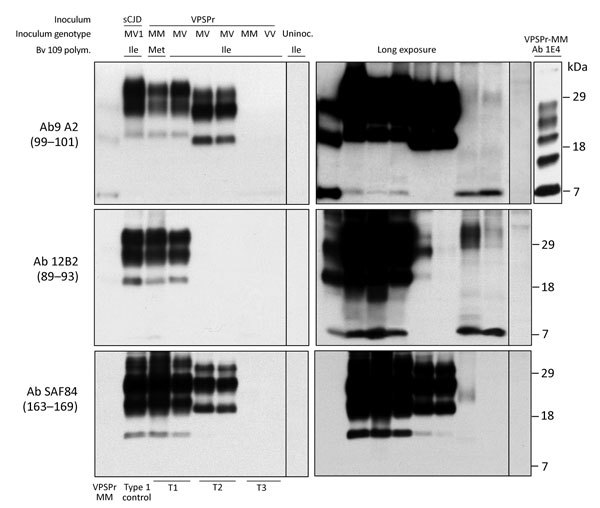
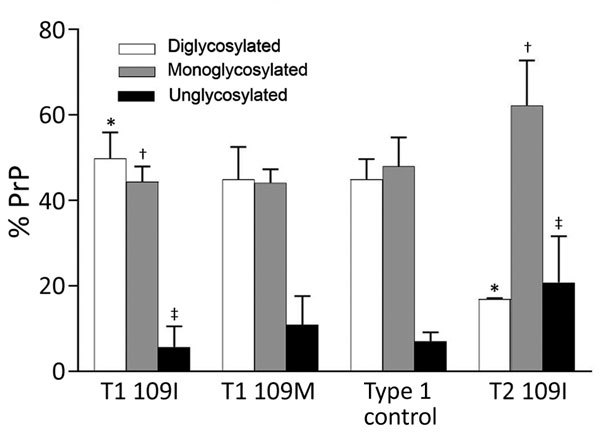
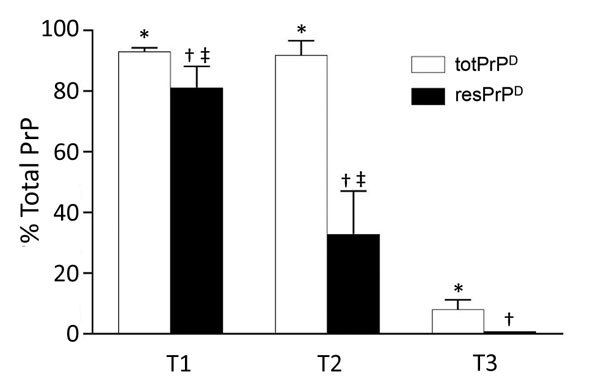
Variably protease-sensitive prionopathy (VPSPr), a recently described human sporadic prion disease, features a protease-resistant, disease-related prion protein (resPrPD) displaying 5 fragments reminiscent of Gerstmann-Sträussler-Scheinker disease. Experimental VPSPr transmission to human PrP-expressing transgenic mice, although replication of the VPSPr resPrPD profile succeeded, has been incomplete because of second passage failure. We bioassayed VPSPr in bank voles, which are susceptible to human prion strains. Transmission was complete; first-passage attack rates were 5%-35%, and second-passage rates reached 100% and survival times were 50% shorter. We observed 3 distinct phenotypes and resPrPD profiles; 2 imitated sporadic Creutzfeldt-Jakob disease resPrPD, and 1 resembled Gerstmann-Sträussler-Scheinker disease resPrPD. The first 2 phenotypes may be related to the presence of minor PrPD components in VPSPr. Full VPSPr transmission confirms permissiveness of bank voles to human prions and suggests that bank vole PrP may efficiently reveal an underrepresented native strain but does not replicate the complex VPSPr PrPD profile.
Keywords: 109I; 109M; 7 kDa; Creutzfeldt-Jakob; Gerstmann-Sträussler-Scheinker; VPSPr; Variable protease-sensitive prionopathy; atypical glycoform; bank voles; lesion profile; minor PrP components; phenotype; prion disease; prions; sporadic; strain; substrain; transmissibility; type 1; type 2.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
