

Pathogenic copy number variants that affect gene expression contribute to genomic burden in cerebral palsy
- PMID: 30564460
- PMCID: PMC6294788
- DOI: 10.1038/s41525-018-0073-4
Pathogenic copy number variants that affect gene expression contribute to genomic burden in cerebral palsy
Erratum in
-
Erratum: Author Correction: Pathogenic copy number variants that affect gene expression contribute to genomic burden in cerebral palsy.NPJ Genom Med. 2019 May 31;4:11. doi: 10.1038/s41525-019-0086-7. eCollection 2019. NPJ Genom Med. 2019. PMID: 31231543 Free PMC article.
Abstract
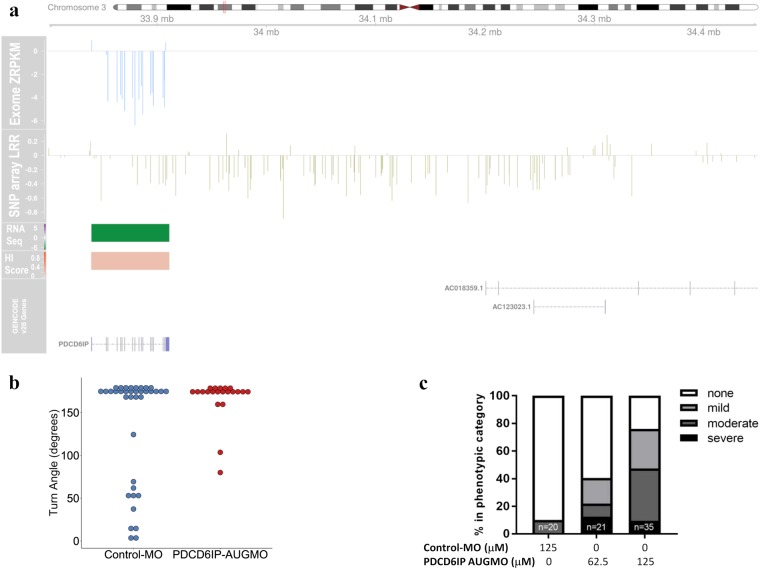
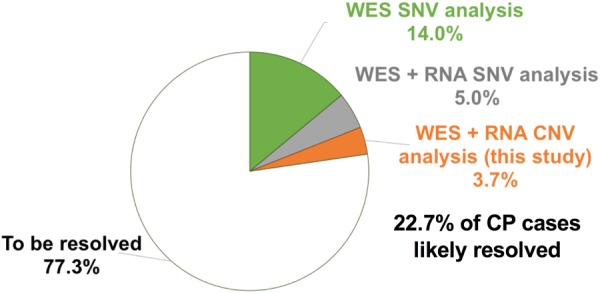
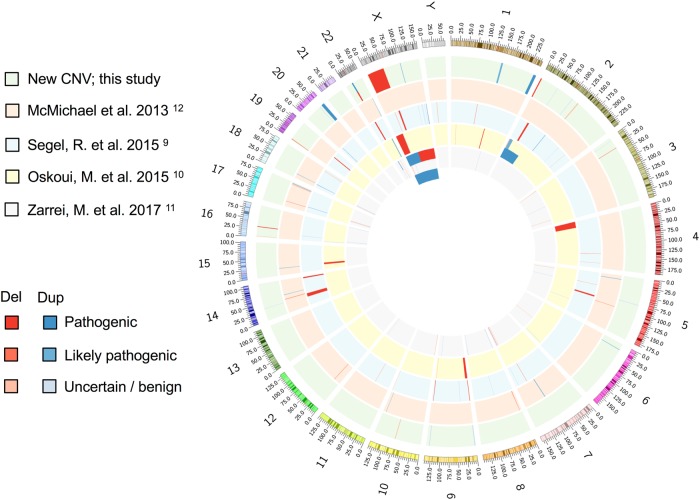
Cerebral palsy (CP) is the most frequent movement disorder of childhood affecting 1 in 500 live births in developed countries. We previously identified likely pathogenic de novo or inherited single nucleotide variants (SNV) in 14% (14/98) of trios by exome sequencing and a further 5% (9/182) from evidence of outlier gene expression using RNA sequencing. Here, we detected copy number variants (CNV) from exomes of 186 unrelated individuals with CP (including our original 98 trios) using the CoNIFER algorithm. CNV were validated with Illumina 850 K SNP arrays and compared with RNA-Seq outlier gene expression analysis from lymphoblastoid cell lines (LCL). Gene expression was highly correlated with gene dosage effect. We resolved an additional 3.7% (7/186) of this cohort with pathogenic or likely pathogenic CNV while a further 7.7% (14/186) had CNV of uncertain significance. We identified recurrent genomic rearrangements previously associated with CP due to 2p25.3 deletion, 22q11.2 deletions and duplications and Xp monosomy. We also discovered a deletion of a single gene, PDCD6IP, and performed additional zebrafish model studies to support its single allele loss in CP aetiology. Combined SNV and CNV analysis revealed pathogenic and likely pathogenic variants in 22.7% of unselected individuals with CP.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Delacy MJ, Reid SM. Australian Cerebral Palsy Register Group. Profile of associated impairments at age 5 years in Australia by cerebral palsy subtype and Gross Motor Function Classification System level for birth years 1996 to 2005. Dev. Med. Child Neurol. 2016;58(Suppl 2):50–56. doi: 10.1111/dmcn.13012. - DOI - PubMed
-
- Lynex CN, et al. Homozygosity for a missense mutation in the 67 kDa isoform of glutamate decarboxylase in a family with autosomal recessive spastic cerebral palsy: parallels with Stiff-Person Syndrome and other movement disorders. BMC Neurol. 2004;4:20. doi: 10.1186/1471-2377-4-20. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
