

Predicting Patient Response to the Antiarrhythmic Mexiletine Based on Genetic Variation
- PMID: 30566038
- PMCID: PMC6588292
- DOI: 10.1161/CIRCRESAHA.118.314050
Predicting Patient Response to the Antiarrhythmic Mexiletine Based on Genetic Variation
Abstract
Rationale: Mutations in the SCN5A gene, encoding the α subunit of the Nav1.5 channel, cause a life-threatening form of cardiac arrhythmia, long QT syndrome type 3 (LQT3). Mexiletine, which is structurally related to the Na+ channel-blocking anesthetic lidocaine, is used to treat LQT3 patients. However, the patient response is variable, depending on the genetic mutation in SCN5A.
Objective: The goal of this study is to understand the molecular basis of patients' variable responses and build a predictive statistical model that can be used to personalize mexiletine treatment based on patient's genetic variant.
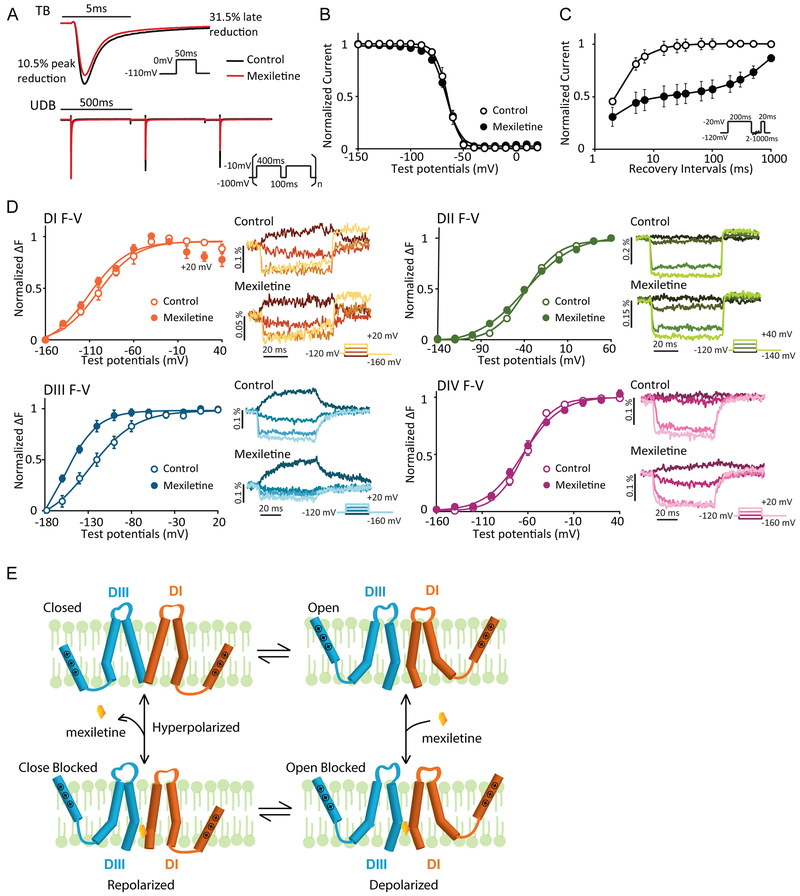
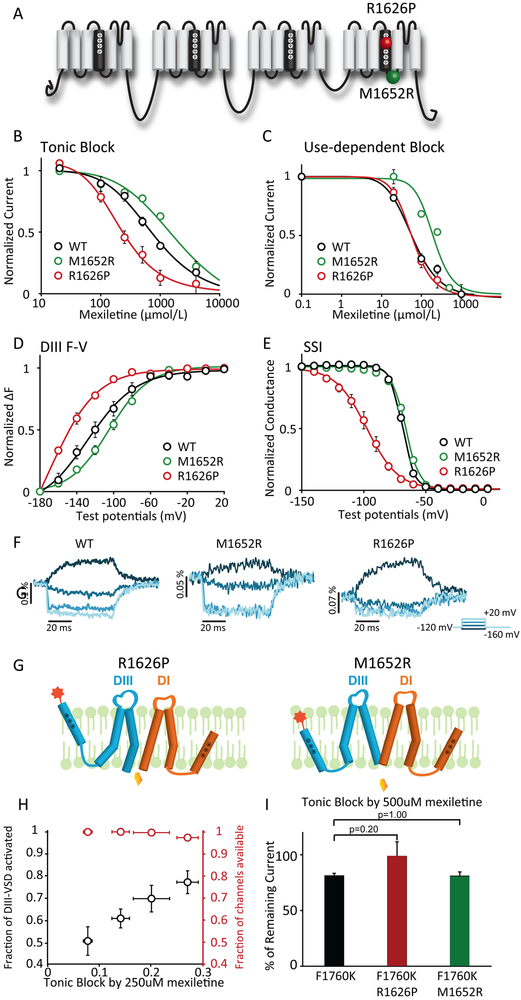
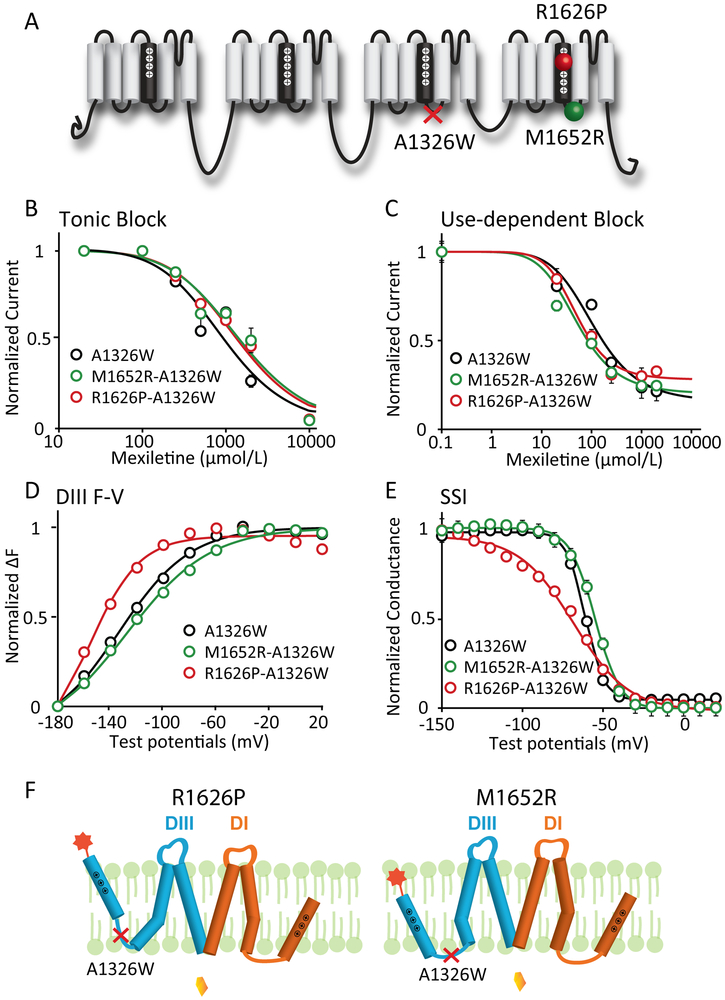
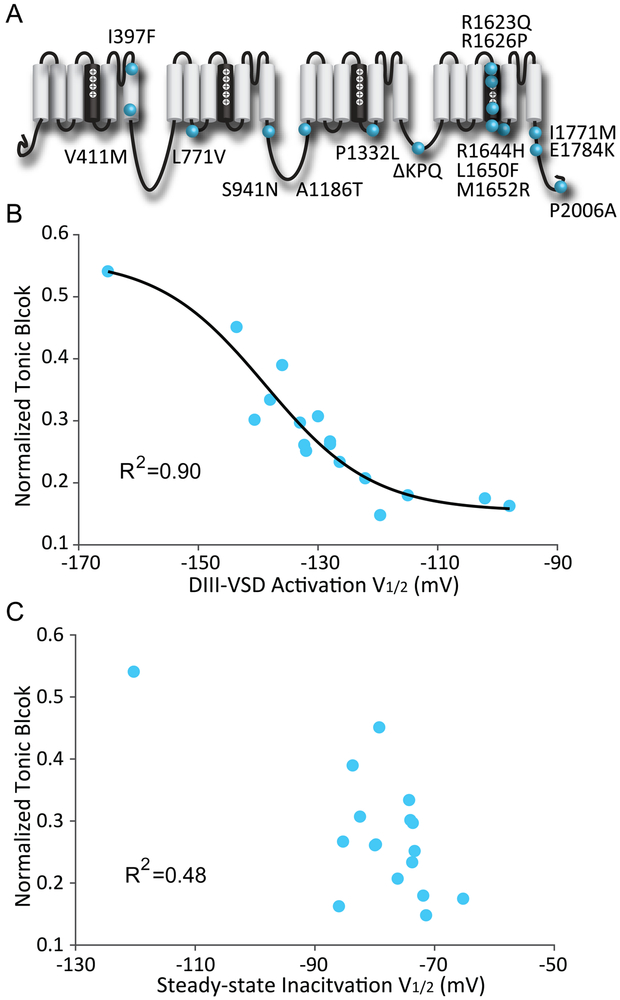
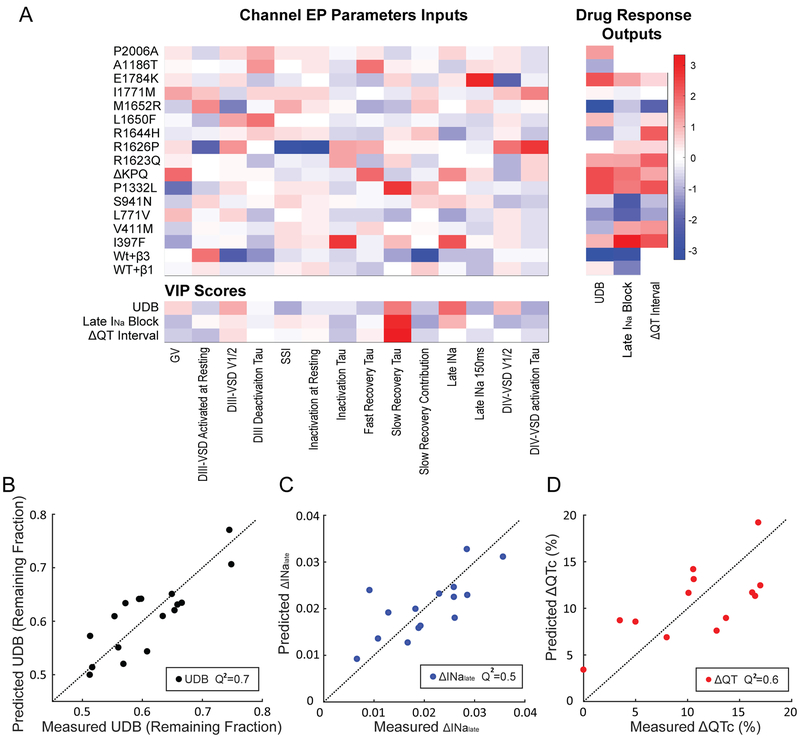
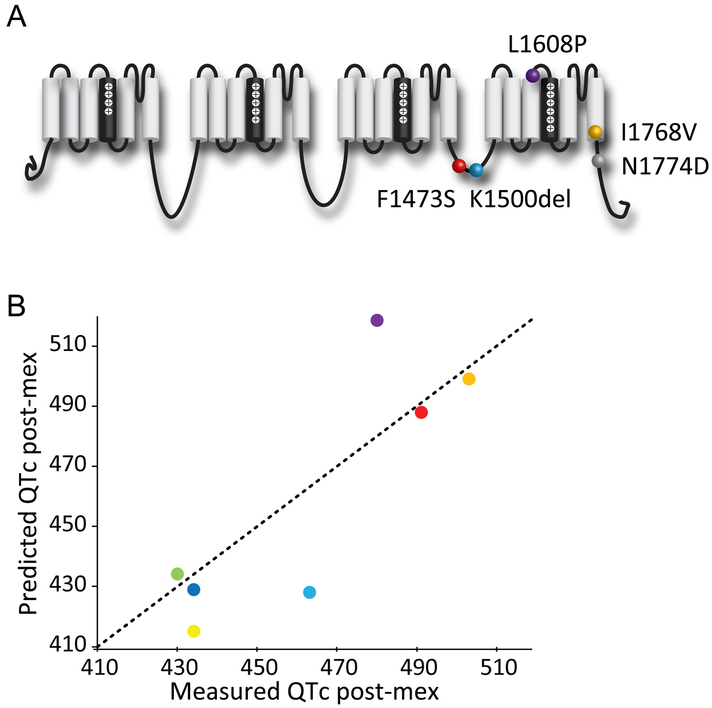
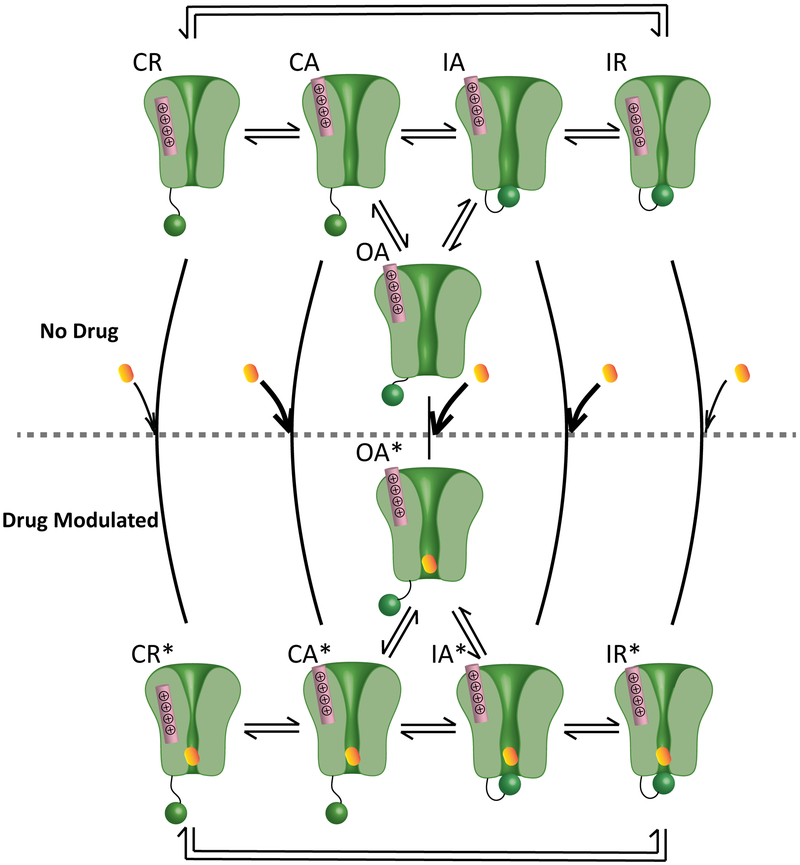
Methods and results: We monitored the cardiac Na+ channel voltage-sensing domain (VSD) conformational dynamics simultaneously with other gating properties for the LQT3 variants. To systematically identify the relationship between mexiletine block and channel biophysical properties, we used a system-based statistical modeling approach to connect the multivariate properties to patient phenotype. We found that mexiletine altered the conformation of the Domain III VSD, which is the same VSD that many tested LQT3 mutations affect. Analysis of 15 LQT3 variants showed a strong correlation between the activation of the Domain III-VSD and the strength of the inhibition of the channel by mexiletine. Based on this improved molecular-level understanding, we generated a systems-based model based on a dataset of 32 LQT3 patients, which then successfully predicted the response of 7 out of 8 patients to mexiletine in a blinded, retrospective trial.
Conclusions: Our results imply that the modulated receptor theory of local anesthetic action, which confines local anesthetic binding effects to the channel pore, should be revised to include drug interaction with the Domain III-VSD. Using an algorithm that incorporates this mode of action, we can predict patient-specific responses to mexiletine, improving therapeutic decision making.
Keywords: electrophysiology; ion channels; long QT syndrome; mexiletine; precision medicine.
Figures
Comment in
-
Precision Versus Traditional Medicine-Clinical Questions Trigger Progress in Basic Science.Circ Res. 2019 Feb 15;124(4):459-461. doi: 10.1161/CIRCRESAHA.119.314629. Circ Res. 2019. PMID: 30763224 No abstract available.
References
-
- Priori SG, Blomström-Lundqvist C, Mazzanti A. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J [Internet]. 2015;8:746–837. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16935866
-
- Zipes DP, Jalife J. Cardiac Electrophysiology: From Cell to Bedside: Sixth Edition. 2013.
-
- Ruan Y, Liu N, Bloise R, Napolitano C, Priori SG. Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation. 2007;116:1137–1144. - PubMed
-
- Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D, Barker AH, Arensberg D, Baker A, Friedman L, Greene HL, Huther ML, Richardson DW. Mortality and Morbidity in Patients Receiving Encainide, Flecainide, or Placebo. N Engl J Med [Internet]. 1991;324:781–788. Available from: http://www.nejm.org/doi/abs/10.1056/NEJM199103213241201 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
