

Unintended specificity of an engineered ligand-binding protein facilitated by unpredicted plasticity of the protein fold
- PMID: 30566669
- PMCID: PMC6692866
- DOI: 10.1093/protein/gzy031
Unintended specificity of an engineered ligand-binding protein facilitated by unpredicted plasticity of the protein fold
Abstract
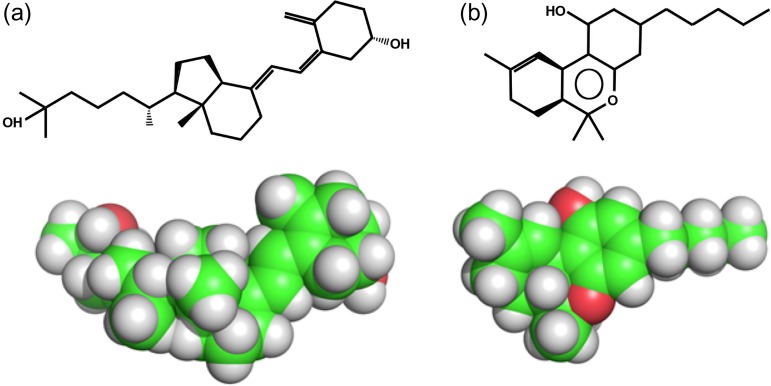
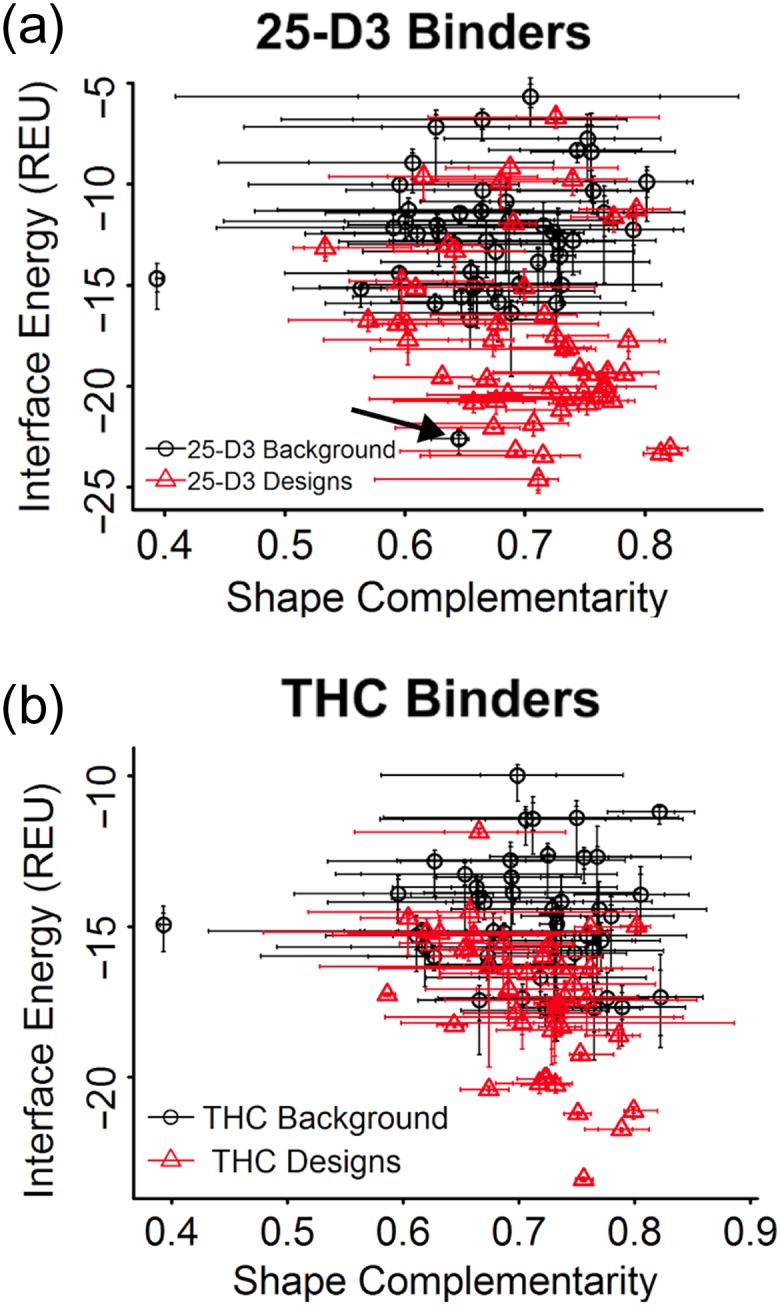
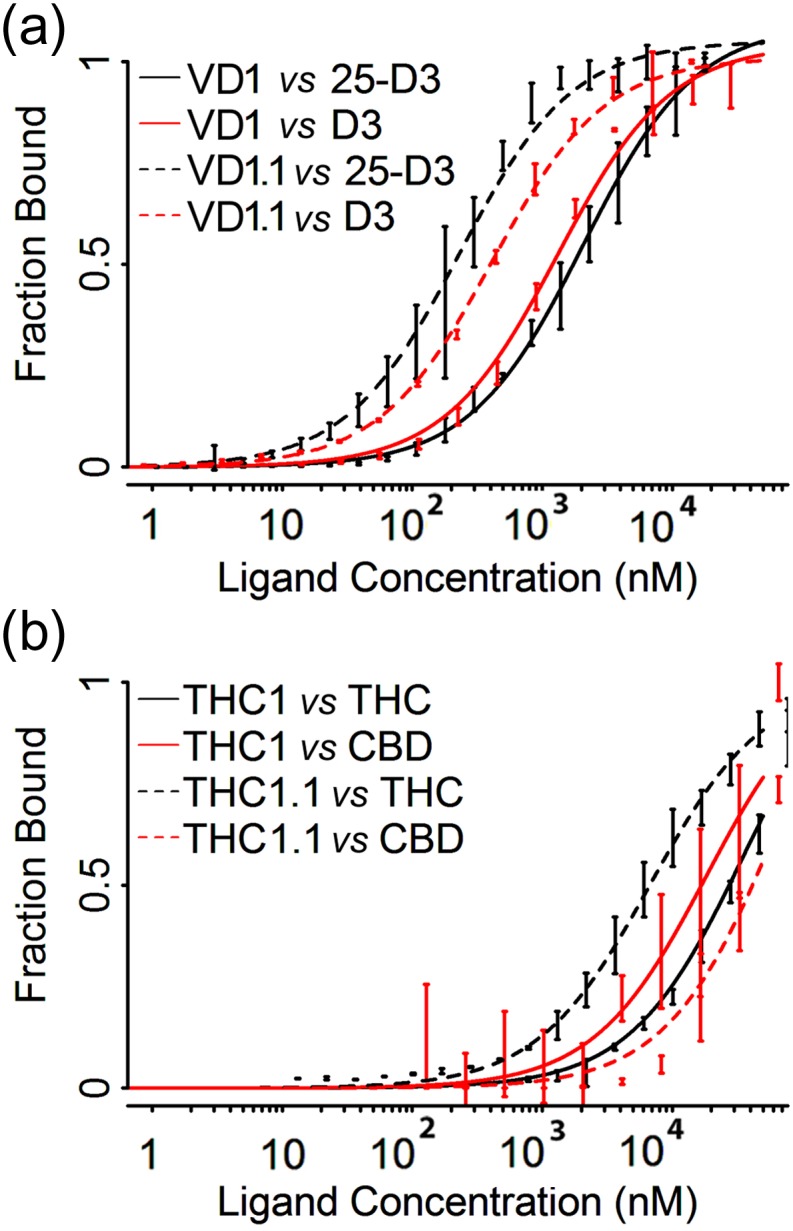
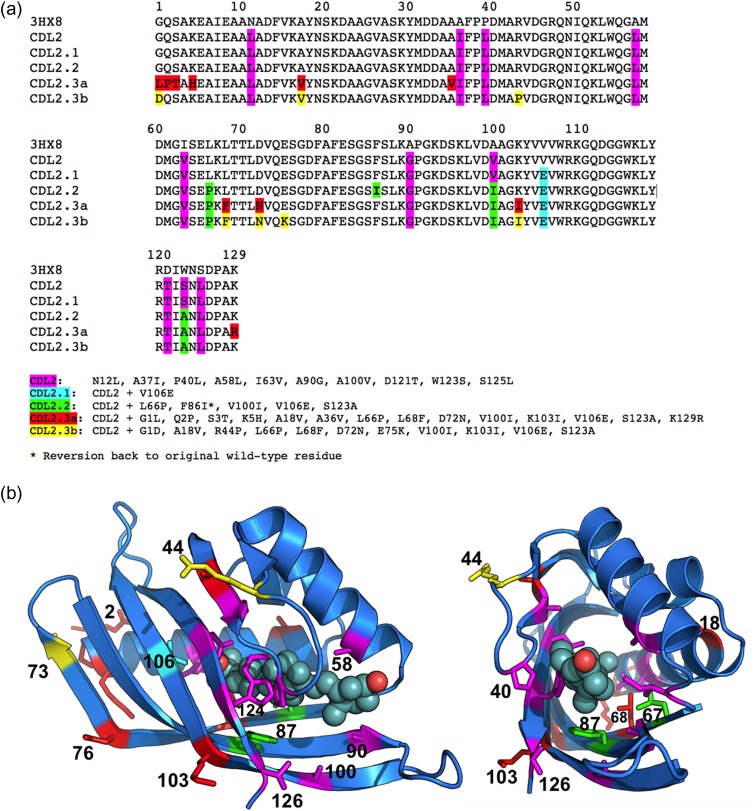
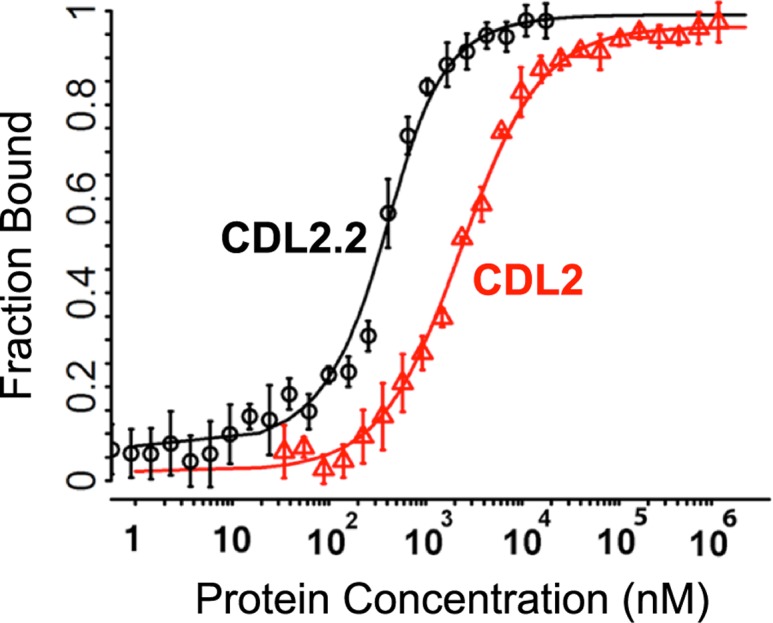
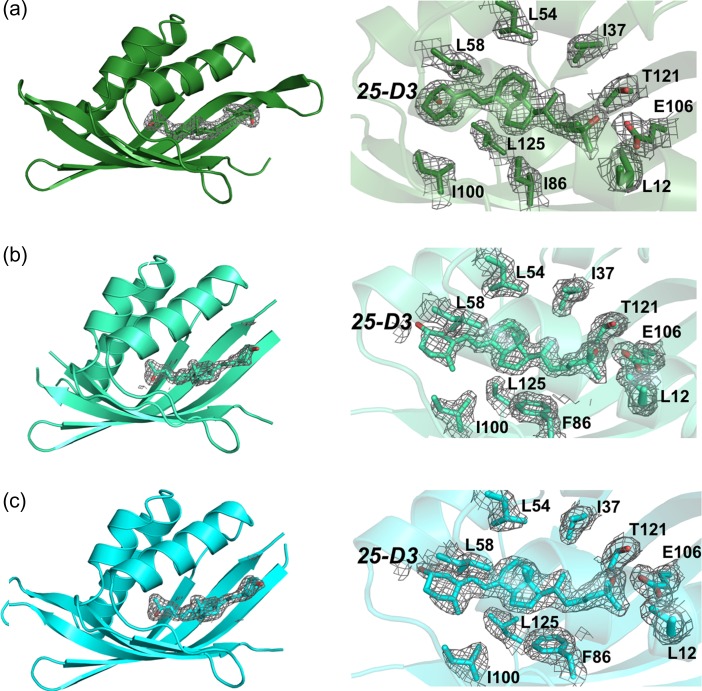
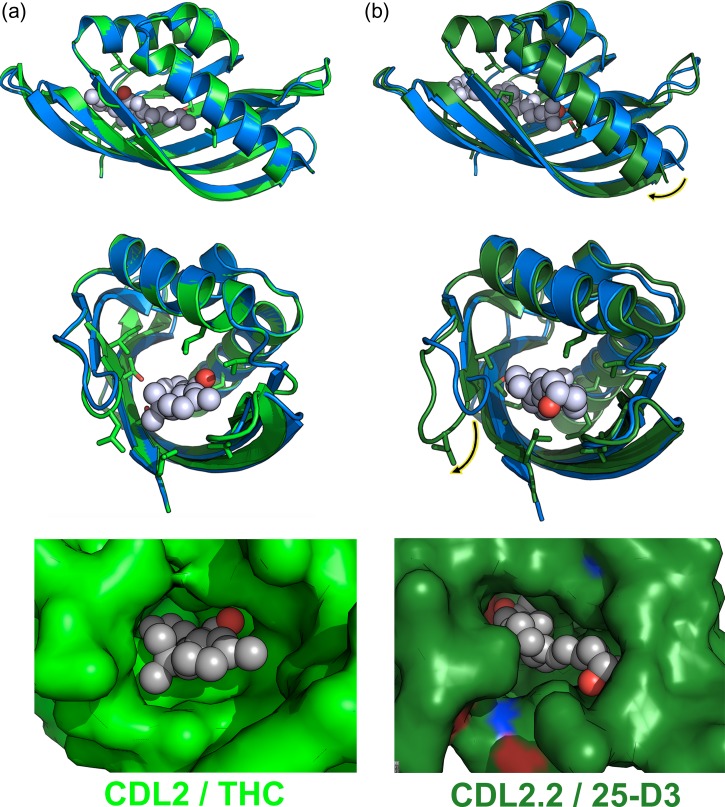
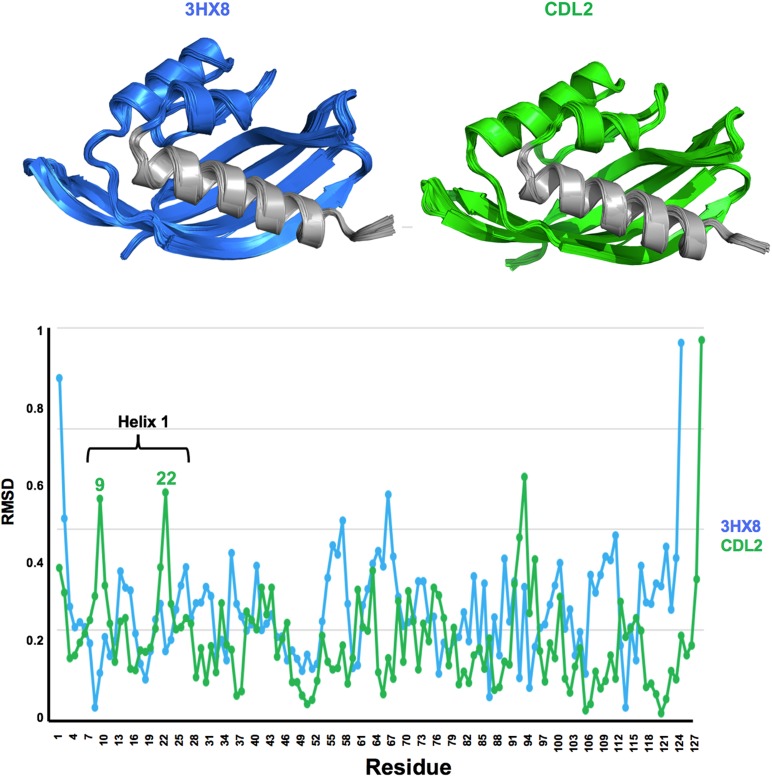
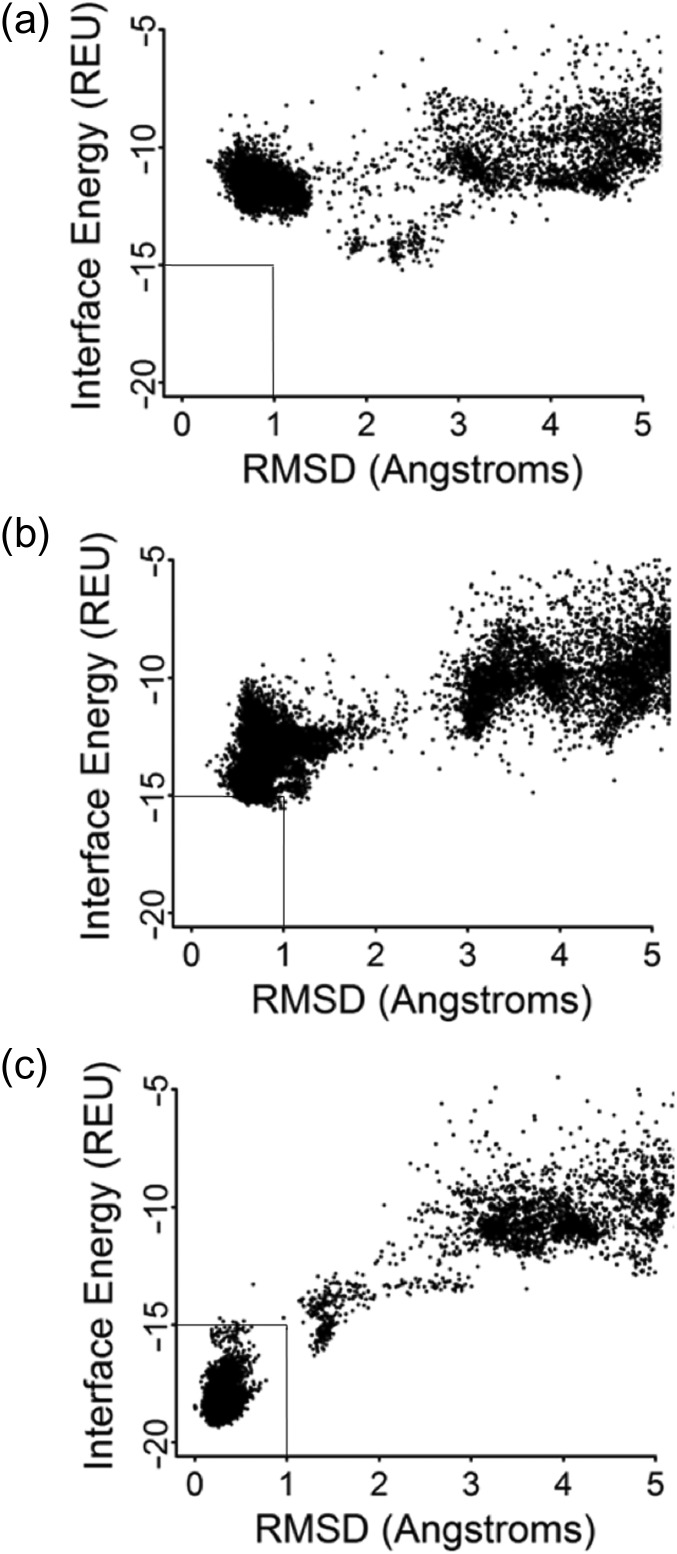
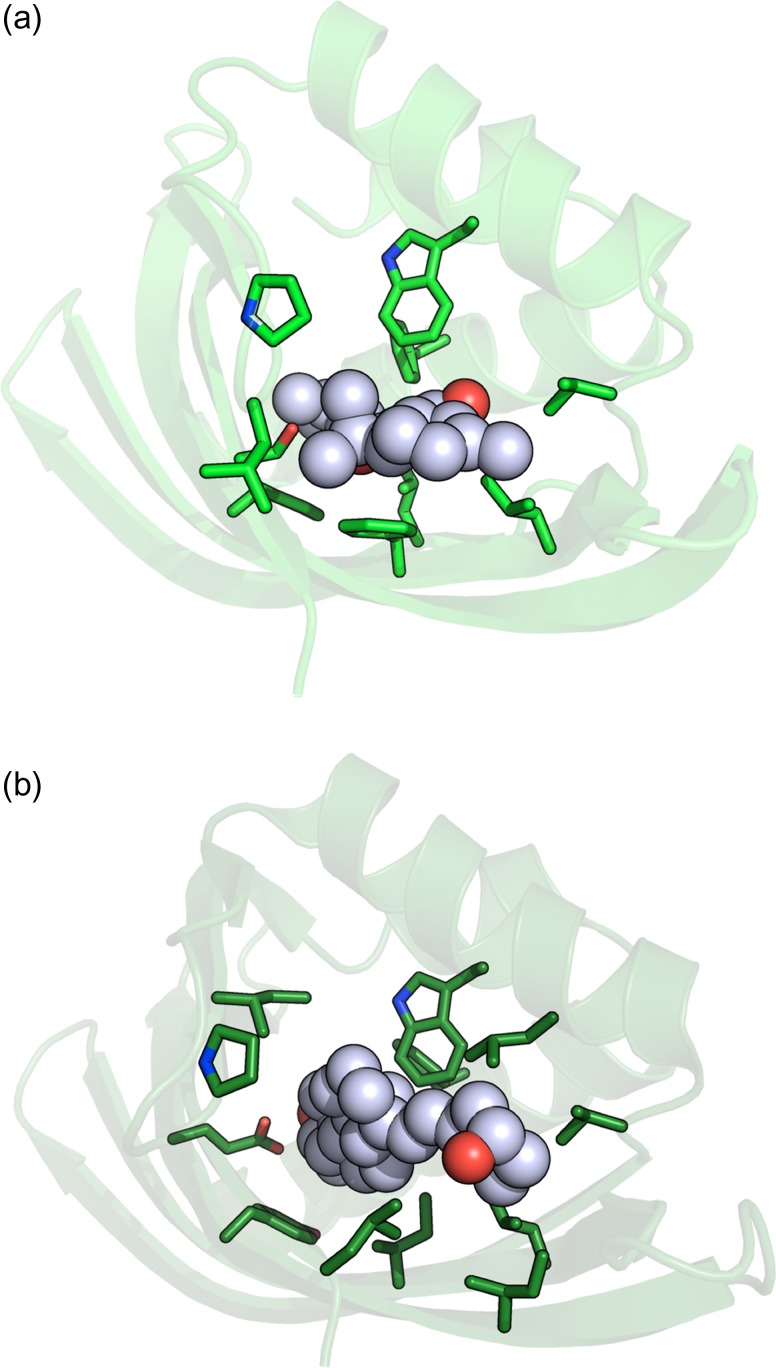
Attempts to create novel ligand-binding proteins often focus on formation of a binding pocket with shape complementarity against the desired ligand (particularly for compounds that lack distinct polar moieties). Although designed proteins often exhibit binding of the desired ligand, in some cases they display unintended recognition behavior. One such designed protein, that was originally intended to bind tetrahydrocannabinol (THC), was found instead to display binding of 25-hydroxy-cholecalciferol (25-D3) and was subjected to biochemical characterization, further selections for enhanced 25-D3 binding affinity and crystallographic analyses. The deviation in specificity is due in part to unexpected altertion of its conformation, corresponding to a significant change of the orientation of an α-helix and an equally large movement of a loop, both of which flank the designed ligand-binding pocket. Those changes led to engineered protein constructs that exhibit significantly more contacts and complementarity towards the 25-D3 ligand than the initial designed protein had been predicted to form towards its intended THC ligand. Molecular dynamics simulations imply that the initial computationally designed mutations may contribute to the movement of the helix. These analyses collectively indicate that accurate prediction and control of backbone dynamics conformation, through a combination of improved conformational sampling and/or de novo structure design, represents a key area of further development for the design and optimization of engineered ligand-binding proteins.
Keywords: affinity versus specificity; crystal structure; ligand binding; protein engineering.
© The Author(s) 2018. Published by Oxford University Press. All rights reserved. For Permissions, please e-mail: journals.permissions@oup.com.
Figures
Similar articles
-
Structural mechanism of specific ligand recognition by a lipocalin tailored for the complexation of digoxigenin.J Mol Biol. 2003 Jul 4;330(2):385-96. doi: 10.1016/s0022-2836(03)00573-4. J Mol Biol. 2003. PMID: 12823976
-
Off-Pocket Activity Cliffs: A Puzzling Facet of Molecular Recognition.J Chem Inf Model. 2020 Jan 27;60(1):152-161. doi: 10.1021/acs.jcim.9b00731. Epub 2019 Dec 16. J Chem Inf Model. 2020. PMID: 31790251
-
Crystal structure of the abl-SH3 domain complexed with a designed high-affinity peptide ligand: implications for SH3-ligand interactions.J Mol Biol. 1998 Aug 21;281(3):513-21. doi: 10.1006/jmbi.1998.1932. J Mol Biol. 1998. PMID: 9698566
-
Computational protein design of ligand binding and catalysis.Curr Opin Chem Biol. 2013 Dec;17(6):929-33. doi: 10.1016/j.cbpa.2013.10.002. Curr Opin Chem Biol. 2013. PMID: 24466576 Review.
-
Alternative binding proteins: anticalins - harnessing the structural plasticity of the lipocalin ligand pocket to engineer novel binding activities.FEBS J. 2008 Jun;275(11):2677-83. doi: 10.1111/j.1742-4658.2008.06439.x. Epub 2008 Apr 24. FEBS J. 2008. PMID: 18435758 Review.
Cited by
-
Advances in the Computational Design of Small-Molecule-Controlled Protein-Based Circuits for Synthetic Biology.Proc IEEE Inst Electr Electron Eng. 2022 May;110(5):659-674. doi: 10.1109/JPROC.2022.3157898. Epub 2022 Apr 8. Proc IEEE Inst Electr Electron Eng. 2022. PMID: 36531560 Free PMC article.
-
THC and CBD: Similarities and differences between siblings.Neuron. 2023 Feb 1;111(3):302-327. doi: 10.1016/j.neuron.2022.12.022. Epub 2023 Jan 12. Neuron. 2023. PMID: 36638804 Free PMC article. Review.
-
Optimization of Protein Thermostability and Exploitation of Recognition Behavior to Engineer Altered Protein-DNA Recognition.Structure. 2020 Jul 7;28(7):760-775.e8. doi: 10.1016/j.str.2020.04.009. Epub 2020 Apr 30. Structure. 2020. PMID: 32359399 Free PMC article.
-
A defined structural unit enables de novo design of small-molecule-binding proteins.Science. 2020 Sep 4;369(6508):1227-1233. doi: 10.1126/science.abb8330. Science. 2020. PMID: 32883865 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
