

Resistance to Second-Generation HIV-1 Maturation Inhibitors
- PMID: 30567982
- PMCID: PMC6401422
- DOI: 10.1128/JVI.02017-18
Resistance to Second-Generation HIV-1 Maturation Inhibitors
Abstract
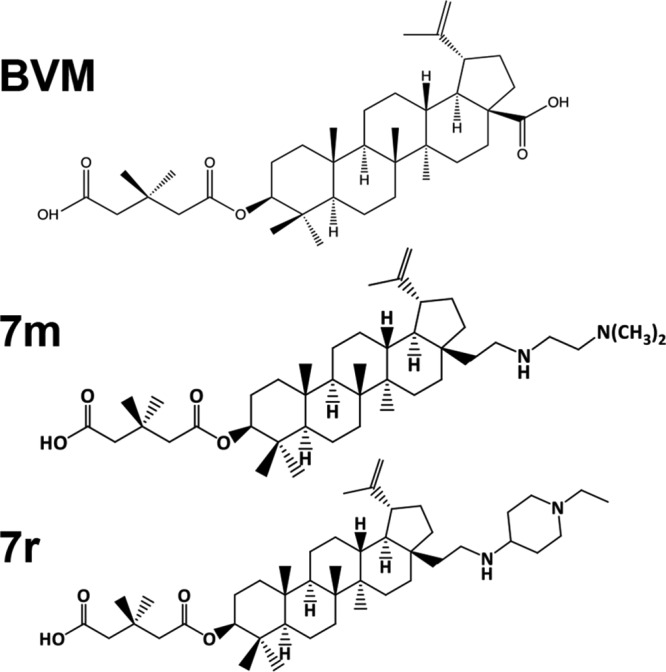
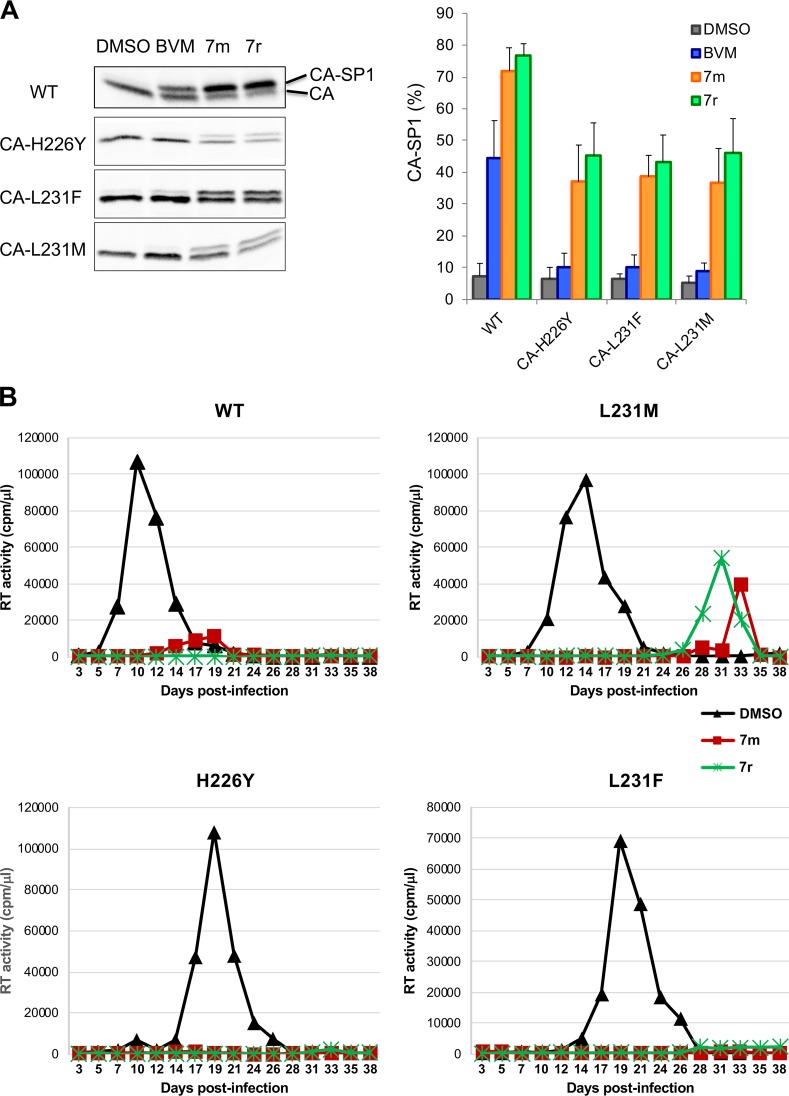
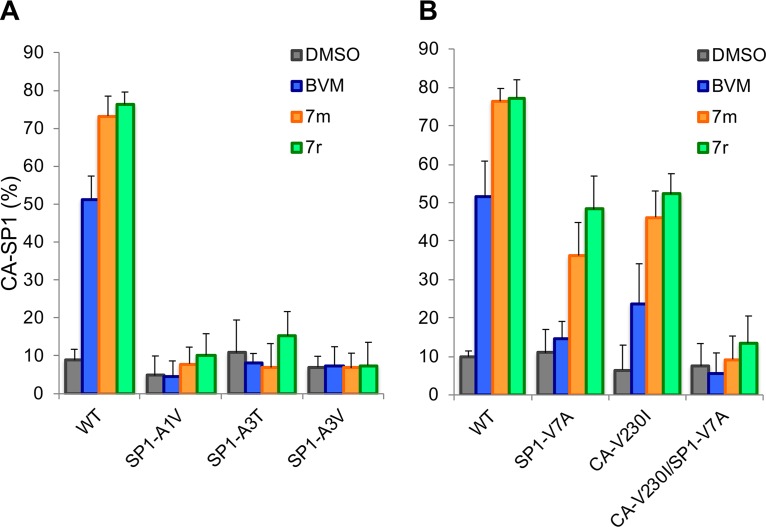
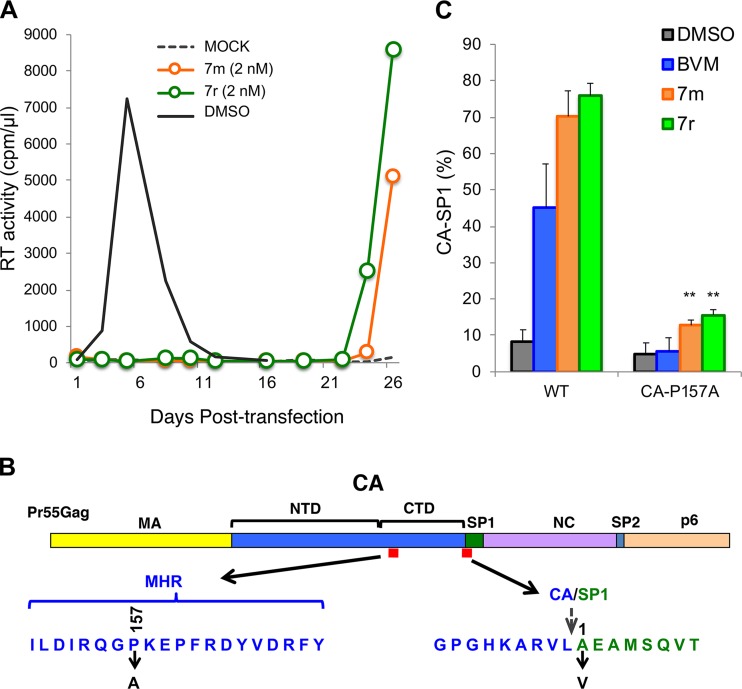
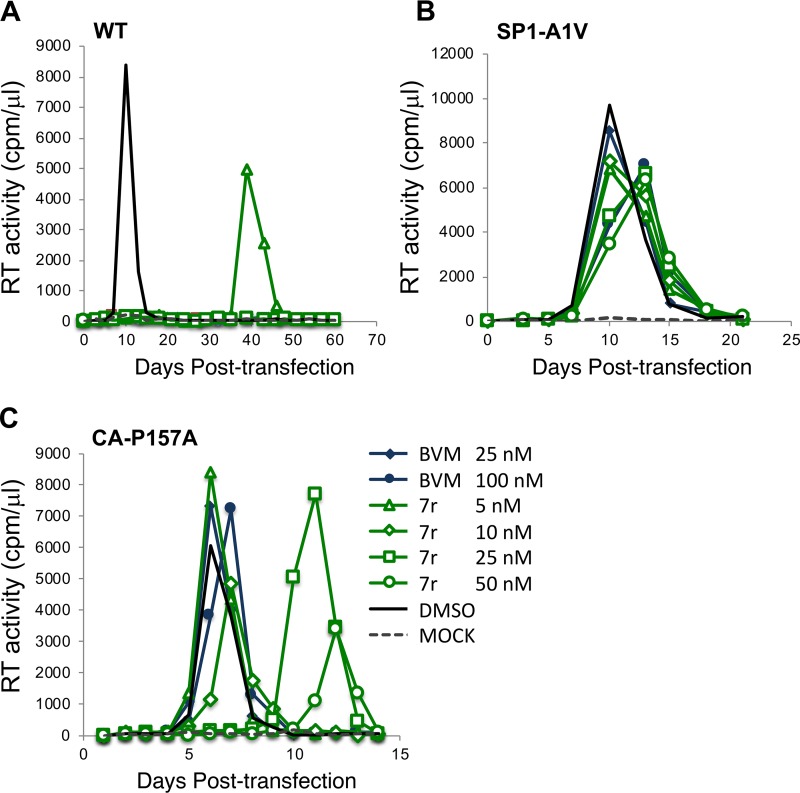
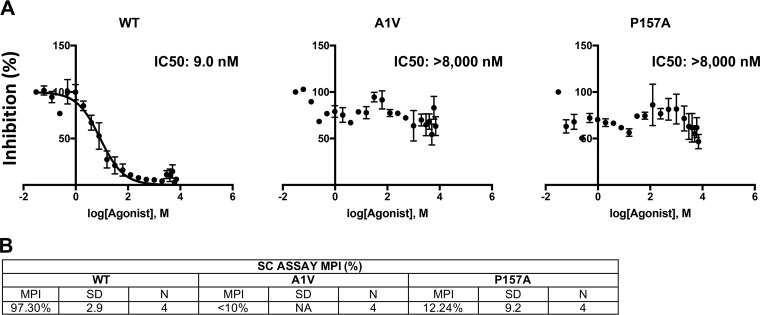
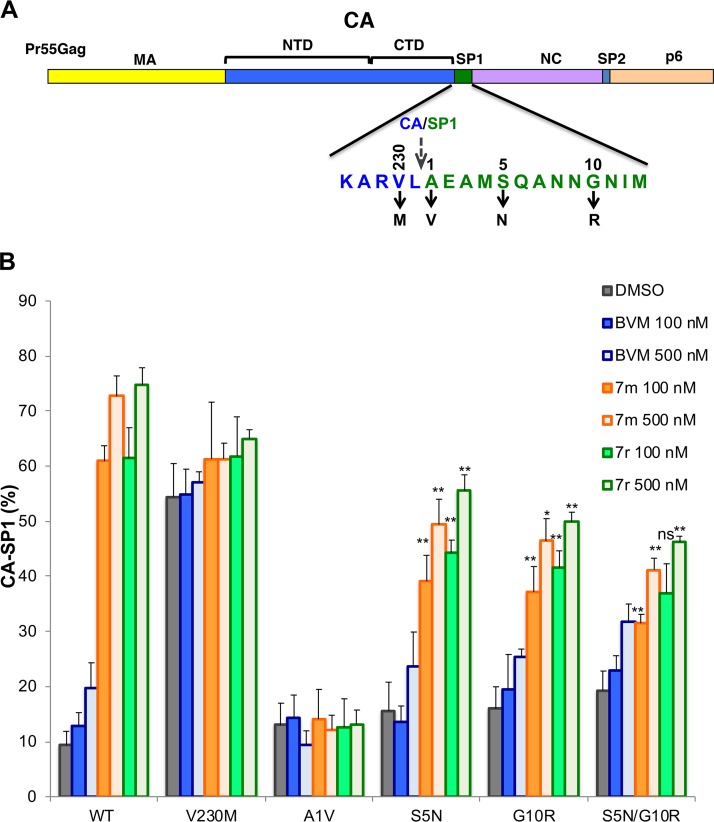
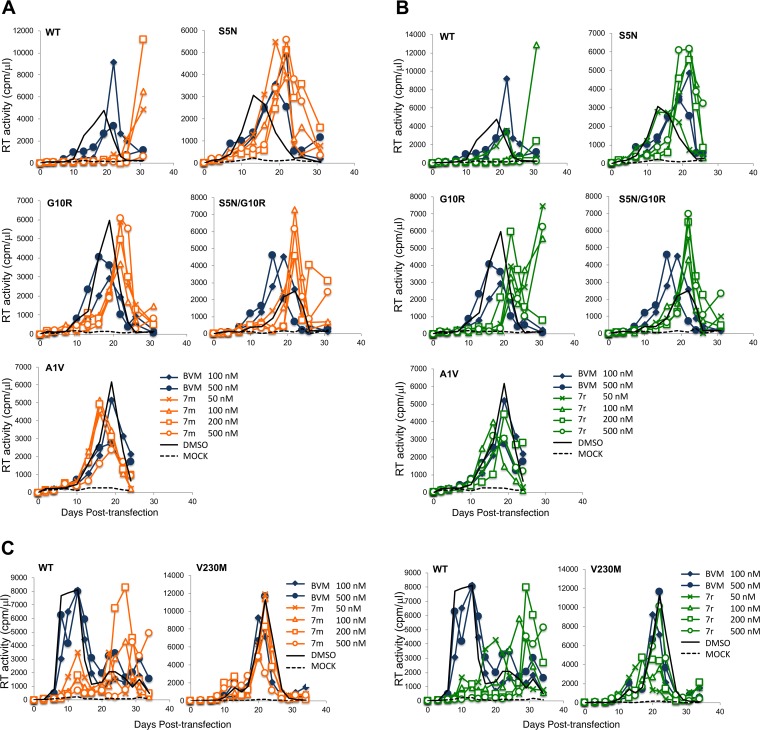
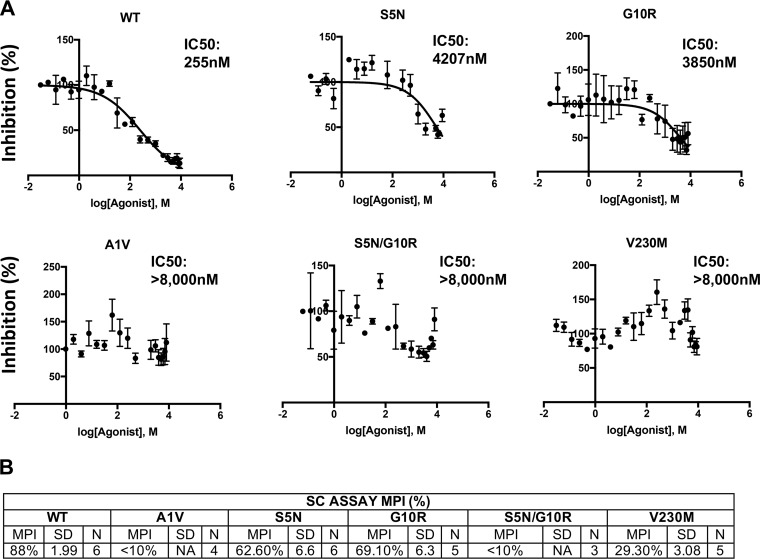
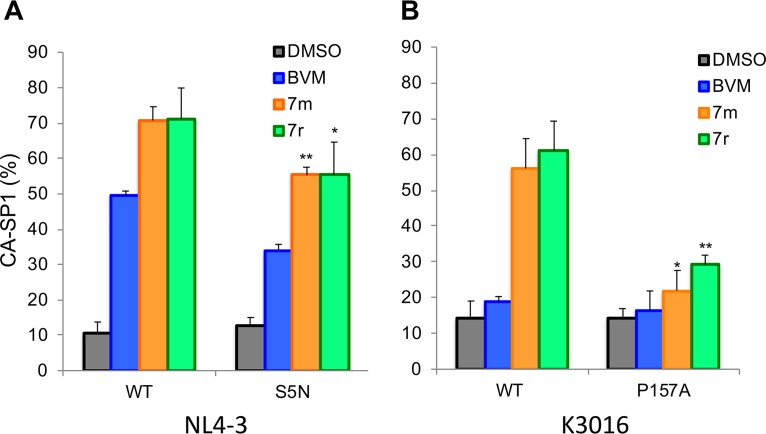
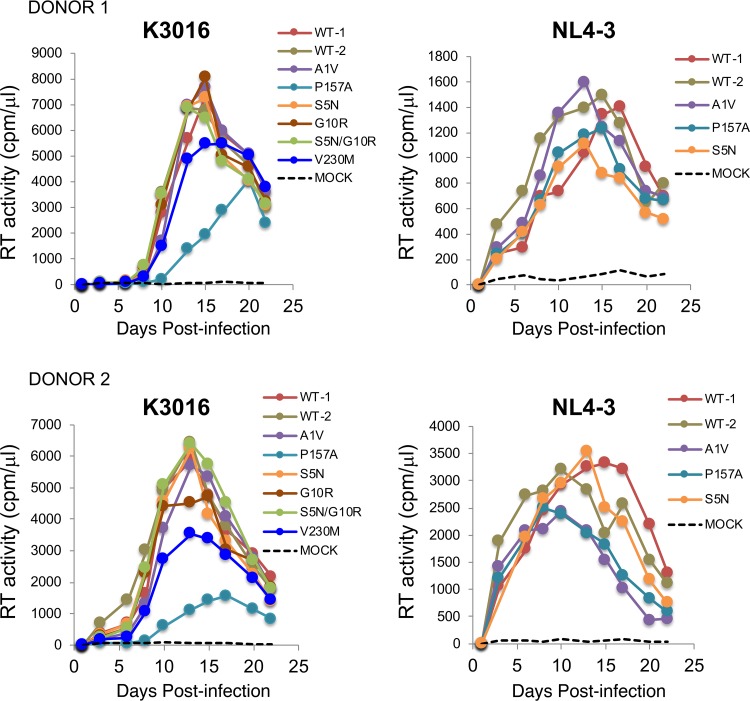
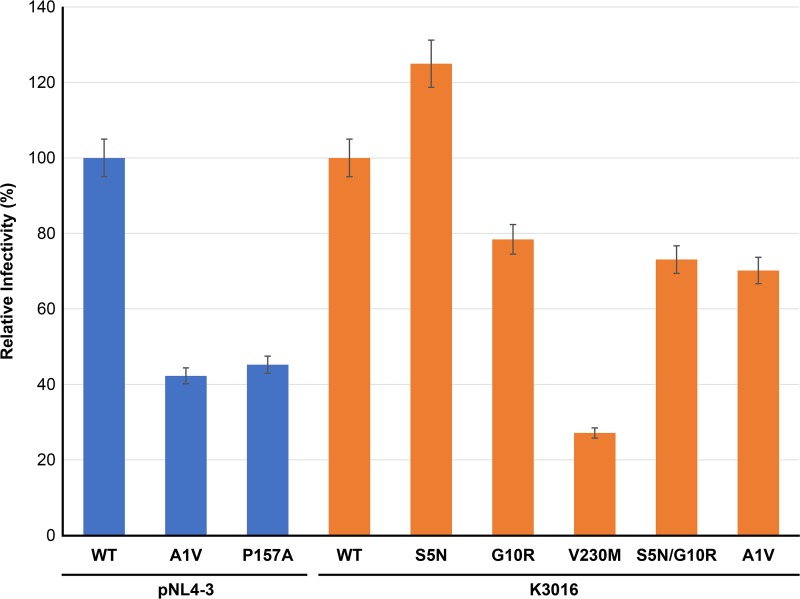
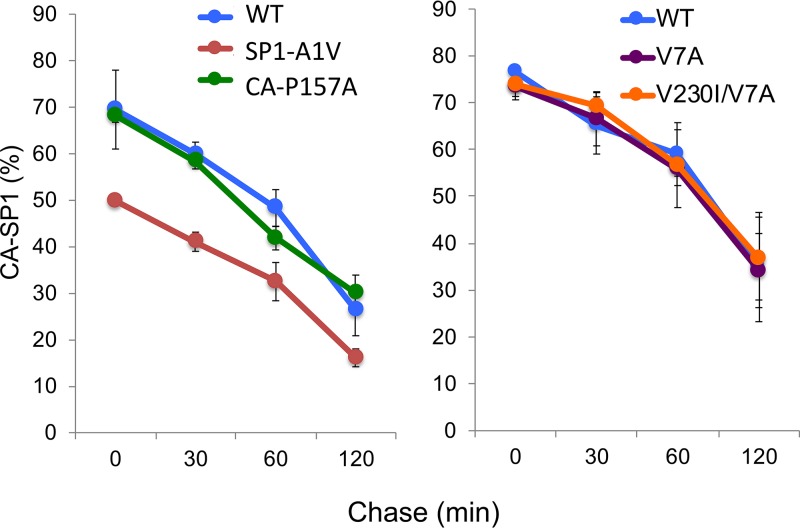
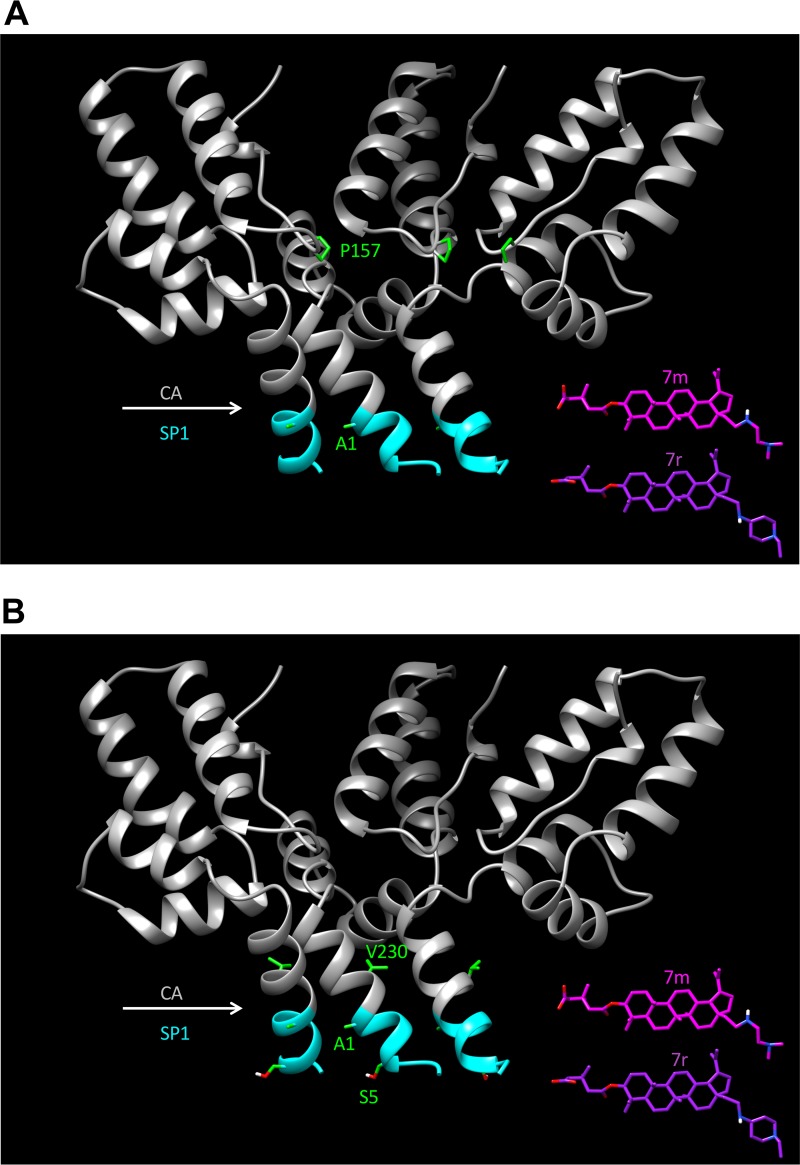
A betulinic acid-based compound, bevirimat (BVM), inhibits HIV-1 maturation by blocking a late step in protease-mediated Gag processing: the cleavage of the capsid-spacer peptide 1 (CA-SP1) intermediate to mature CA. Previous studies showed that mutations conferring resistance to BVM cluster around the CA-SP1 cleavage site. Single amino acid polymorphisms in the SP1 region of Gag and the C terminus of CA reduced HIV-1 susceptibility to BVM, leading to the discontinuation of BVM's clinical development. We recently reported a series of "second-generation" BVM analogs that display markedly improved potency and breadth of activity relative to the parent molecule. Here, we demonstrate that viral clones bearing BVM resistance mutations near the C terminus of CA are potently inhibited by second-generation BVM analogs. We performed de novo selection experiments to identify mutations that confer resistance to these novel compounds. Selection experiments with subtype B HIV-1 identified an Ala-to-Val mutation at SP1 residue 1 and a Pro-to-Ala mutation at CA residue 157 within the major homology region (MHR). In selection experiments with subtype C HIV-1, we identified mutations at CA residue 230 (CA-V230M) and SP1 residue 1 (SP1-A1V), residue 5 (SP1-S5N), and residue 10 (SP1-G10R). The positions at which resistance mutations arose are highly conserved across multiple subtypes of HIV-1. We demonstrate that the mutations confer modest to high-level maturation inhibitor resistance. In most cases, resistance was not associated with a detectable increase in the kinetics of CA-SP1 processing. These results identify mutations that confer resistance to second-generation maturation inhibitors and provide novel insights into the mechanism of resistance.IMPORTANCE HIV-1 maturation inhibitors are a class of small-molecule compounds that block a late step in the viral protease-mediated processing of the Gag polyprotein precursor, the viral protein responsible for the formation of virus particles. The first-in-class HIV-1 maturation inhibitor bevirimat was highly effective in blocking HIV-1 replication, but its activity was compromised by naturally occurring sequence polymorphisms within Gag. Recently developed bevirimat analogs, referred to as "second-generation" maturation inhibitors, overcome this issue. To understand more about how these second-generation compounds block HIV-1 maturation, here we selected for HIV-1 mutants that are resistant to these compounds. Selections were performed in the context of two different subtypes of HIV-1. We identified a small set of mutations at highly conserved positions within the capsid and spacer peptide 1 domains of Gag that confer resistance. Identification and analysis of these maturation inhibitor-resistant mutants provide insights into the mechanisms of resistance to these compounds.
Keywords: antiretroviral; human immunodeficiency virus; retrovirus; virus assembly; virus maturation.
Copyright © 2019 American Society for Microbiology.
Figures
References
-
- Guichet E, Aghokeng A, Serrano L, Bado G, Toure-Kane C, Eymard-Duvernay S, Villabona-Arenas CJ, Delaporte E, Ciaffi L, Peeters M. 2016. Short communication: high viral load and multidrug resistance due to late switch to second-line regimens could be a major obstacle to reach the 90-90-90 UNAIDS objectives in sub-Saharan Africa. AIDS Res Hum Retroviruses 32:1159–1162. doi: 10.1089/AID.2016.0010. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
