Identification and characterization of Coronaviridae genomes from Vietnamese bats and rats based on conserved protein domains
- PMID: 30568804
- PMCID: PMC6295324
- DOI: 10.1093/ve/vey035
Identification and characterization of Coronaviridae genomes from Vietnamese bats and rats based on conserved protein domains
Abstract
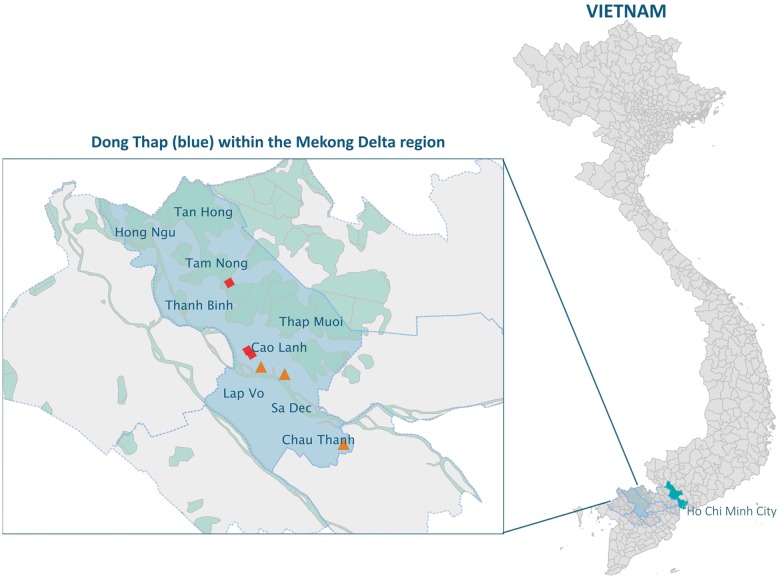
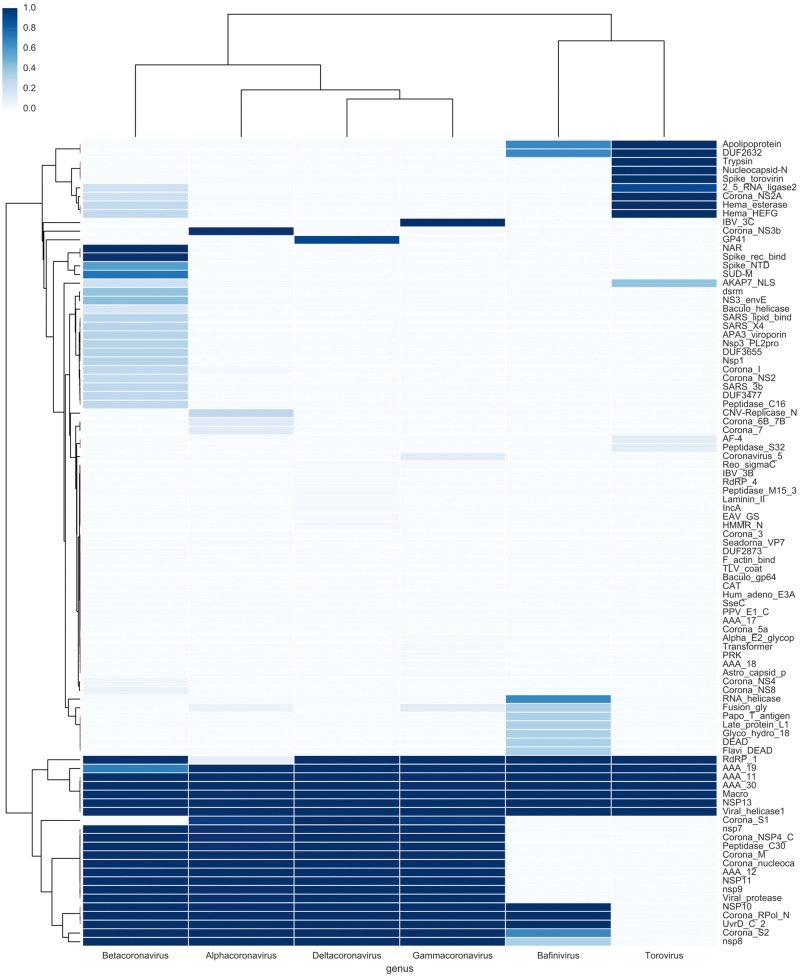
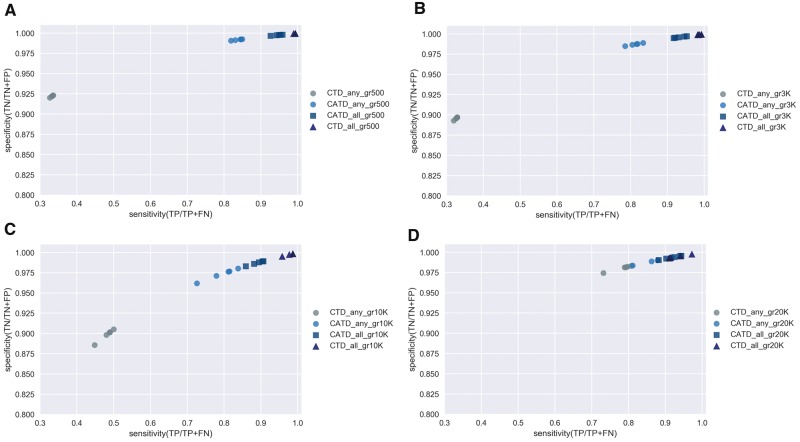
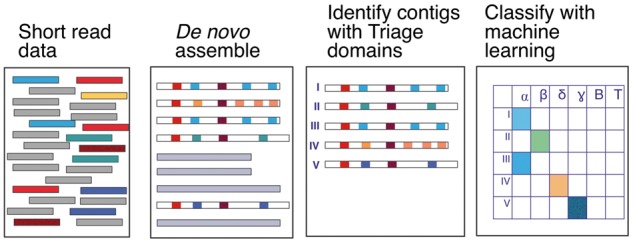
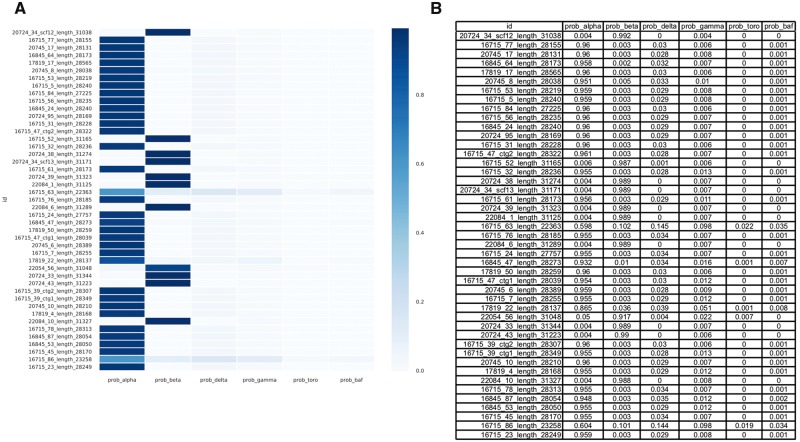
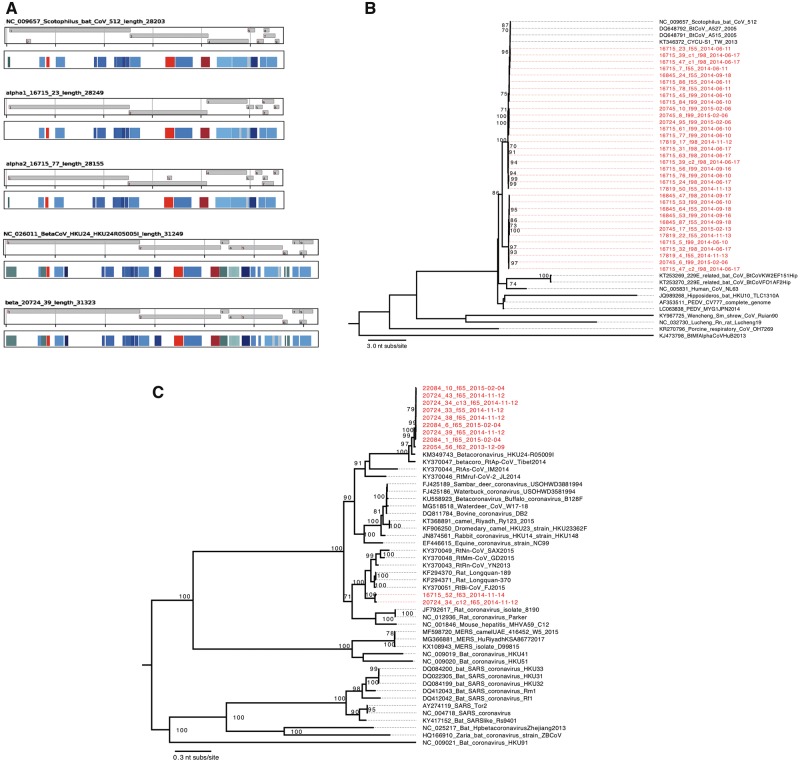
The Coronaviridae family of viruses encompasses a group of pathogens with a zoonotic potential as observed from previous outbreaks of the severe acute respiratory syndrome coronavirus and Middle East respiratory syndrome coronavirus. Accordingly, it seems important to identify and document the coronaviruses in animal reservoirs, many of which are uncharacterized and potentially missed by more standard diagnostic assays. A combination of sensitive deep sequencing technology and computational algorithms is essential for virus surveillance, especially for characterizing novel- or distantly related virus strains. Here, we explore the use of profile Hidden Markov Model-defined Pfam protein domains (Pfam domains) encoded by new sequences as a Coronaviridae sequence classification tool. The encoded domains are used first in a triage to identify potential Coronaviridae sequences and then processed using a Random Forest method to classify the sequences to the Coronaviridae genus level. The application of this algorithm on Coronaviridae genomes assembled from agnostic deep sequencing data from surveillance of bats and rats in Dong Thap province (Vietnam) identified thirty-four Alphacoronavirus and eleven Betacoronavirus genomes. This collection of bat and rat coronaviruses genomes provided essential information on the local diversity of coronaviruses and substantially expanded the number of coronavirus full genomes available from bat and rats and may facilitate further molecular studies on this group of viruses.
Keywords: Pfam; machine learning; profile Hidden Markov model; protein domains; random forest; virus classification.
Figures