

RIP-MD: a tool to study residue interaction networks in protein molecular dynamics
- PMID: 30568854
- PMCID: PMC6287582
- DOI: 10.7717/peerj.5998
RIP-MD: a tool to study residue interaction networks in protein molecular dynamics
Abstract
Protein structure is not static; residues undergo conformational rearrangements and, in doing so, create, stabilize or break non-covalent interactions. Molecular dynamics (MD) is a technique used to simulate these movements with atomic resolution. However, given the data-intensive nature of the technique, gathering relevant information from MD simulations is a complex and time consuming process requiring several computational tools to perform these analyses. Among different approaches, the study of residue interaction networks (RINs) has proven to facilitate the study of protein structures. In a RIN, nodes represent amino-acid residues and the connections between them depict non-covalent interactions. Here, we describe residue interaction networks in protein molecular dynamics (RIP-MD), a visual molecular dynamics (VMD) plugin to facilitate the study of RINs using trajectories obtained from MD simulations of proteins. Our software generates RINs from MD trajectory files. The non-covalent interactions defined by RIP-MD include H-bonds, salt bridges, VdWs, cation-π, π-π, Arginine-Arginine, and Coulomb interactions. In addition, RIP-MD also computes interactions based on distances between Cαs and disulfide bridges. The results of the analysis are shown in an user friendly interface. Moreover, the user can take advantage of the VMD visualization capacities, whereby through some effortless steps, it is possible to select and visualize interactions described for a single, several or all residues in a MD trajectory. Network and descriptive table files are also generated, allowing their further study in other specialized platforms. Our method was written in python in a parallelized fashion. This characteristic allows the analysis of large systems impossible to handle otherwise. RIP-MD is available at http://www.dlab.cl/ripmd.
Keywords: Molecular dynamics; Residue interaction networks; Trajectory analysis; VMD plugin.
Conflict of interest statement
Tomas Perez-Acle is an Academic Editor for PeerJ.
Figures
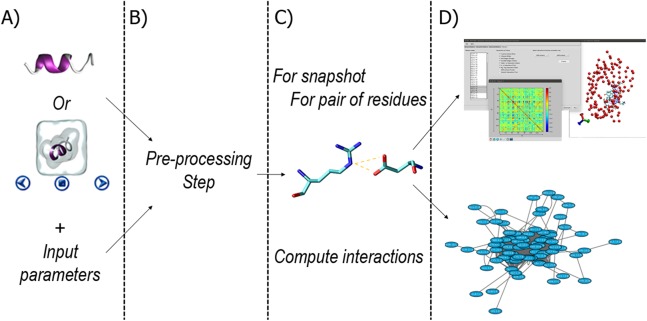
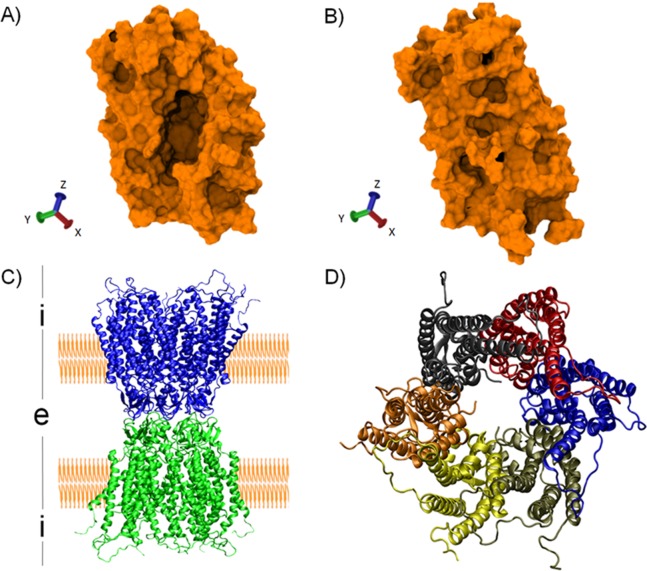
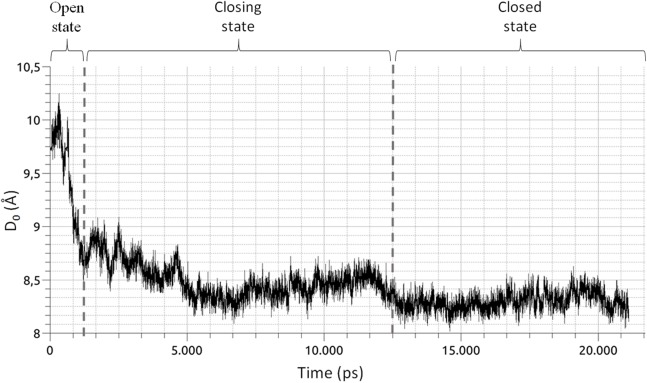
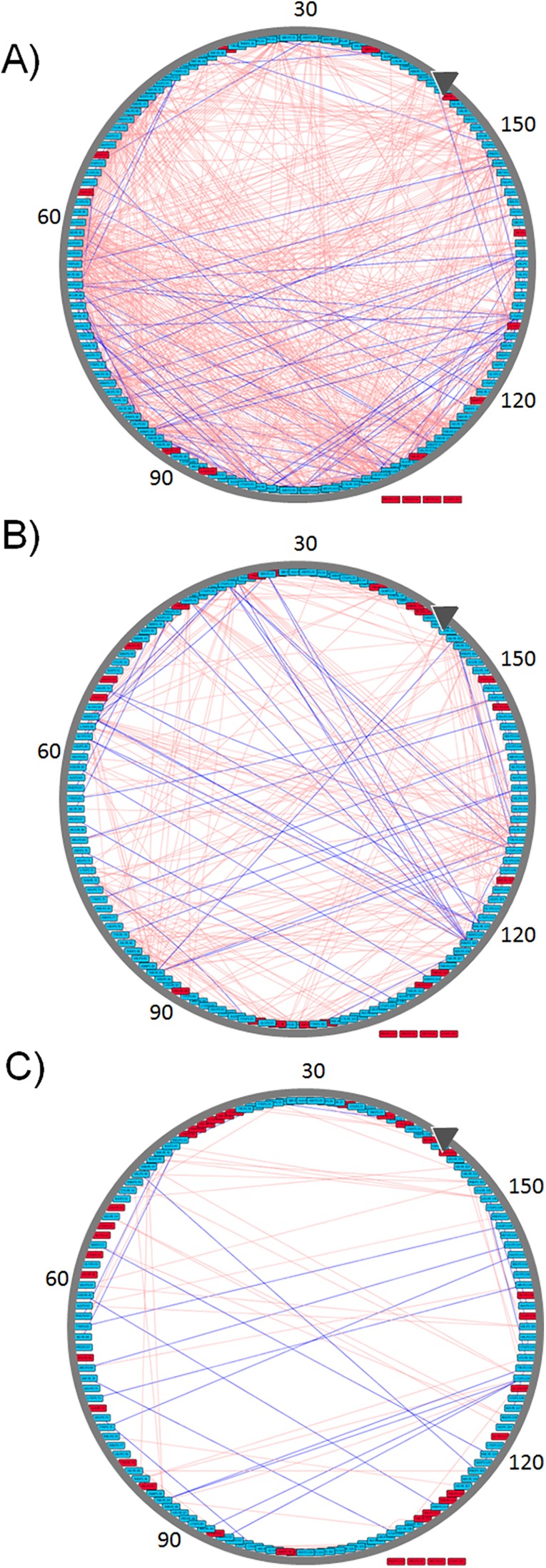
References
-
- Araya-Secchi R, Perez-Acle T, Gu Kang S, Huynh T, Bernardin A, Escalona Y, Garate J-A, Martínez AD, García IE, Sáez JC, Zhou R. Characterization of a novel water pocket inside the human cx26 hemichannel structure. Biophysical Journal. 2014;107(3):599–612. doi: 10.1016/j.bpj.2014.05.037. - DOI - PMC - PubMed
-
- Brandes U. On variants of shortest-path betweenness centrality and their generic computation. Social Networks. 2008;30(2):136–145. doi: 10.1016/j.socnet.2007.11.001. - DOI
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
