

Identification of regulatory elements from nascent transcription using dREG
- PMID: 30573452
- PMCID: PMC6360809
- DOI: 10.1101/gr.238279.118
Identification of regulatory elements from nascent transcription using dREG
Abstract
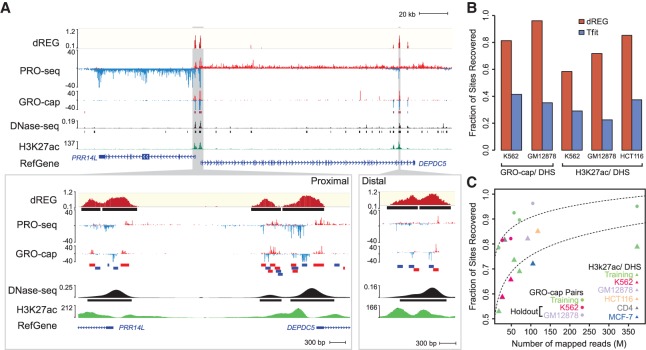
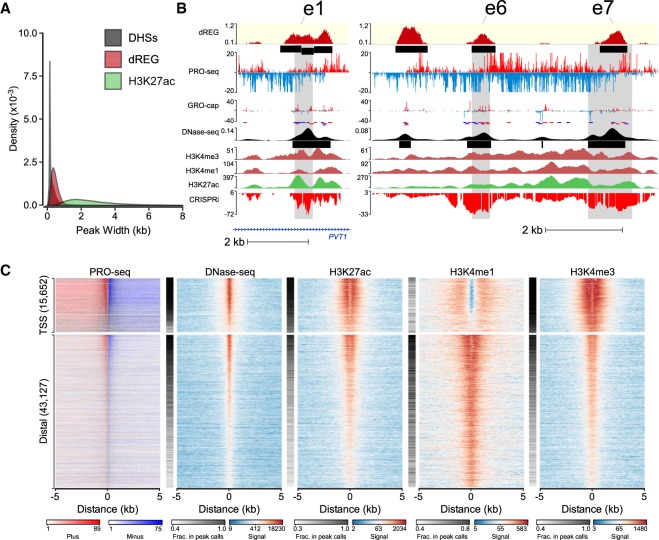
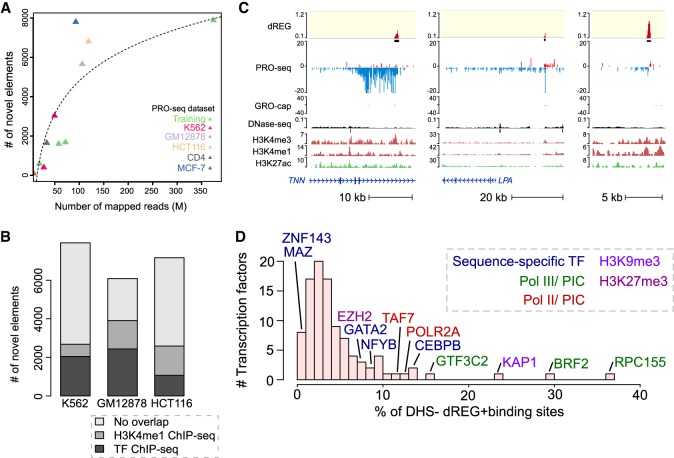
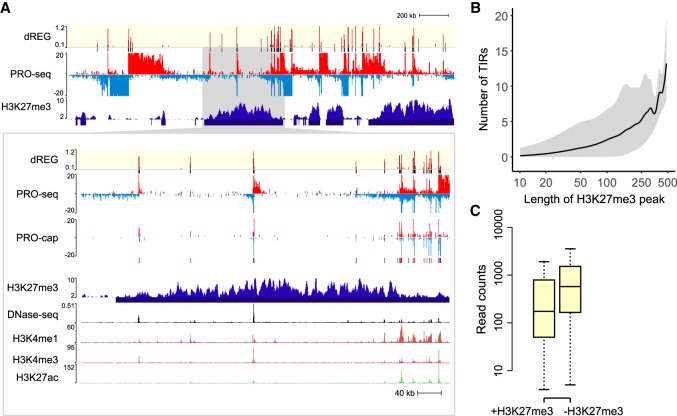
Our genomes encode a wealth of transcription initiation regions (TIRs) that can be identified by their distinctive patterns of actively elongating RNA polymerase. We previously introduced dREG to identify TIRs using PRO-seq data. Here, we introduce an efficient new implementation of dREG that uses PRO-seq data to identify both uni- and bidirectionally transcribed TIRs with 70% improvement in accuracy, three- to fourfold higher resolution, and >100-fold increases in computational efficiency. Using a novel strategy to identify TIRs based on their statistical confidence reveals extensive overlap with orthogonal assays, yet also reveals thousands of additional weakly transcribed TIRs that were not identified by H3K27ac ChIP-seq or DNase-seq. Novel TIRs discovered by dREG were often associated with RNA polymerase III initiation, bound by pioneer transcription factors, or located in broad domains marked by repressive chromatin modifications. Our results suggest that transcription initiation can be a powerful tool for expanding the catalog of functional elements.
© 2019 Wang et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous