

Concise total syntheses of (-)-jorunnamycin A and (-)-jorumycin enabled by asymmetric catalysis
- PMID: 30573544
- PMCID: PMC7017906
- DOI: 10.1126/science.aav3421
Concise total syntheses of (-)-jorunnamycin A and (-)-jorumycin enabled by asymmetric catalysis
Abstract
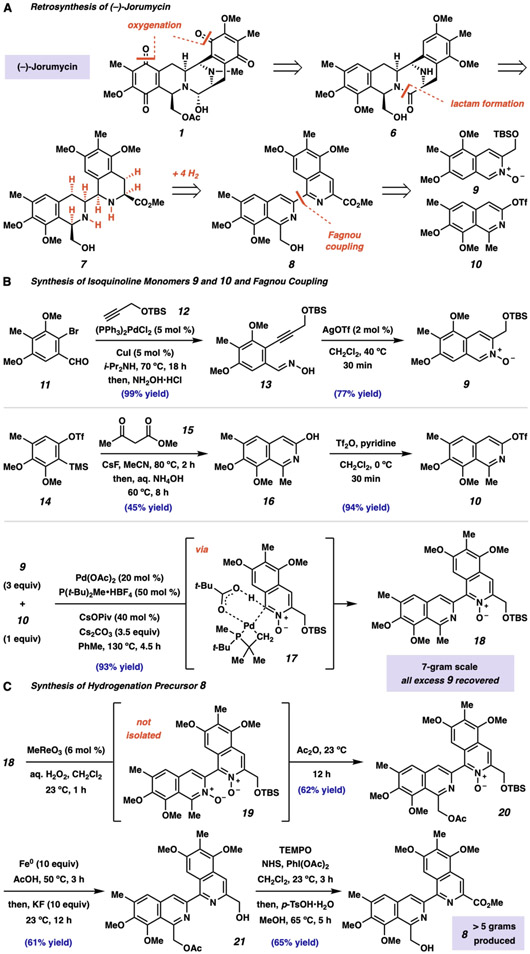
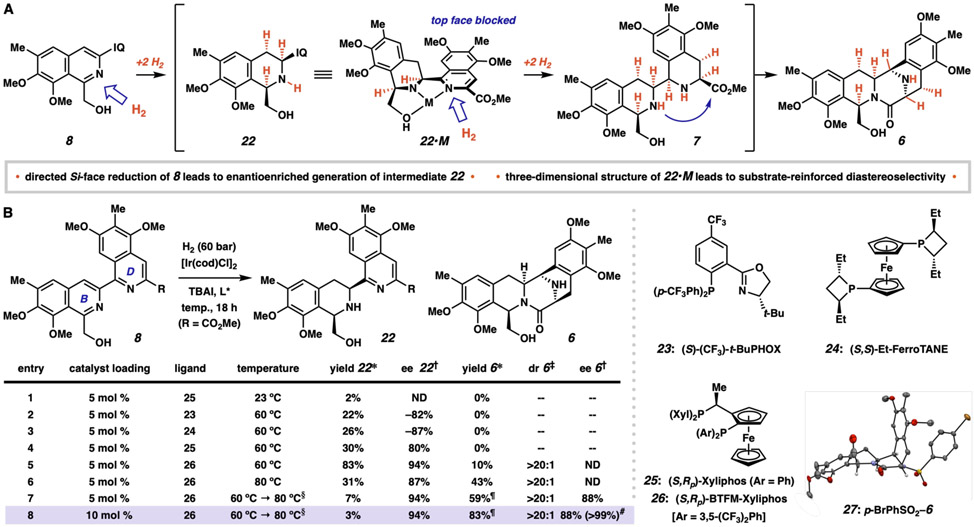
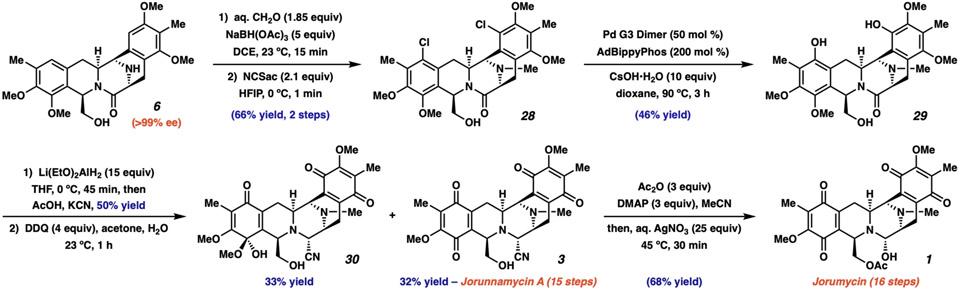
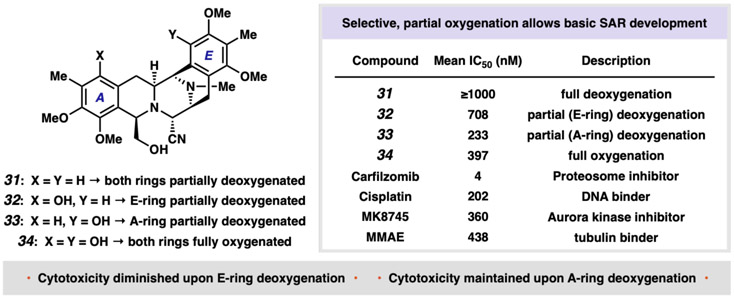
The bis-tetrahydroisoquinoline (bis-THIQ) natural products have been studied intensively over the past four decades for their exceptionally potent anticancer activity, in addition to strong Gram-positive and Gram-negative antibiotic character. Synthetic strategies toward these complex polycyclic compounds have relied heavily on electrophilic aromatic chemistry, such as the Pictet-Spengler reaction, that mimics their biosynthetic pathways. Herein, we report an approach to two bis-THIQ natural products, jorunnamycin A and jorumycin, that instead harnesses the power of modern transition-metal catalysis for the three major bond-forming events and proceeds with high efficiency (15 and 16 steps, respectively). By breaking from biomimicry, this strategy allows for the preparation of a more diverse set of nonnatural analogs.
Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures

References
-
- Chrzanowska M, Grajewska A, Rozwadowska MD, Chem. Rev 116, 12369–12465 (2016). - PubMed
-
- Newman DJ, Cragg GM, J. Nat. Prod 79, 629–661 (2016). - PubMed
-
- Cuevas C, Francesch A, Nat. Prod. Rep 26, 322–337 (2009). - PubMed
-
- Cuevas C, et al. , Org. Lett 2, 2545–2548 (2000). - PubMed
-
- Lown JW, Joshua AV, Lee JS, Biochemistry 21, 419–428 (1982). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
