

Sonic hedgehog signaling instigates high-fat diet-induced insulin resistance by targeting PPARγ stability
- PMID: 30573683
- PMCID: PMC6398147
- DOI: 10.1074/jbc.RA118.004411
Sonic hedgehog signaling instigates high-fat diet-induced insulin resistance by targeting PPARγ stability
Abstract
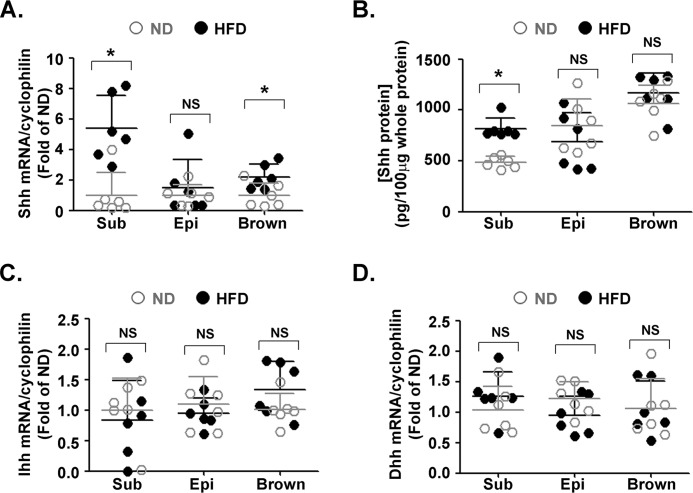
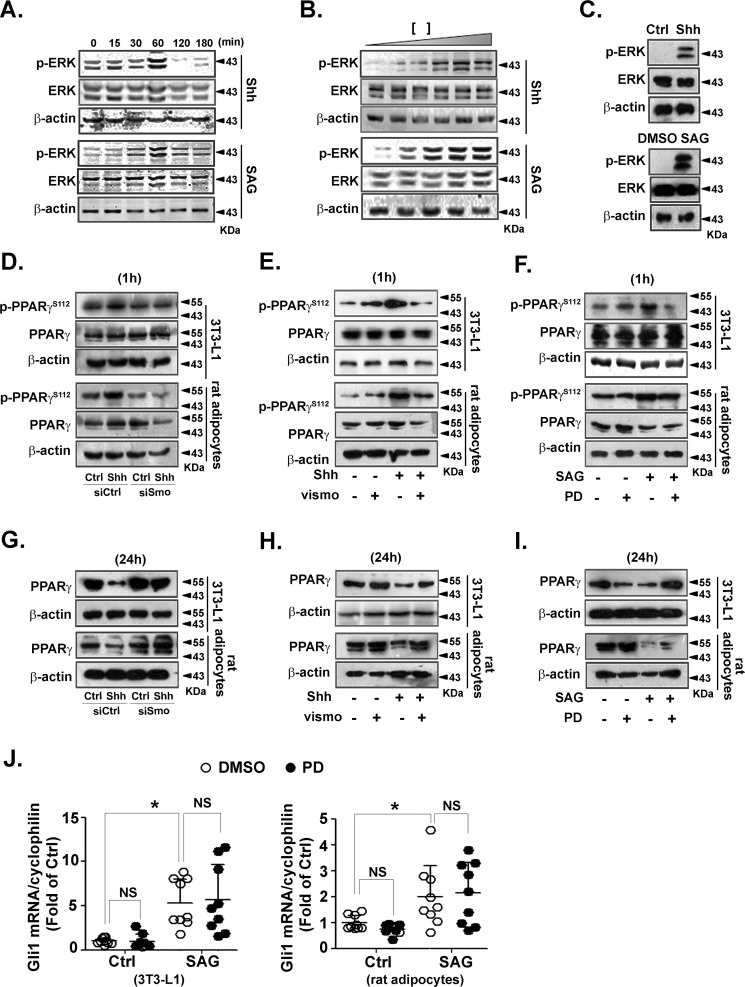
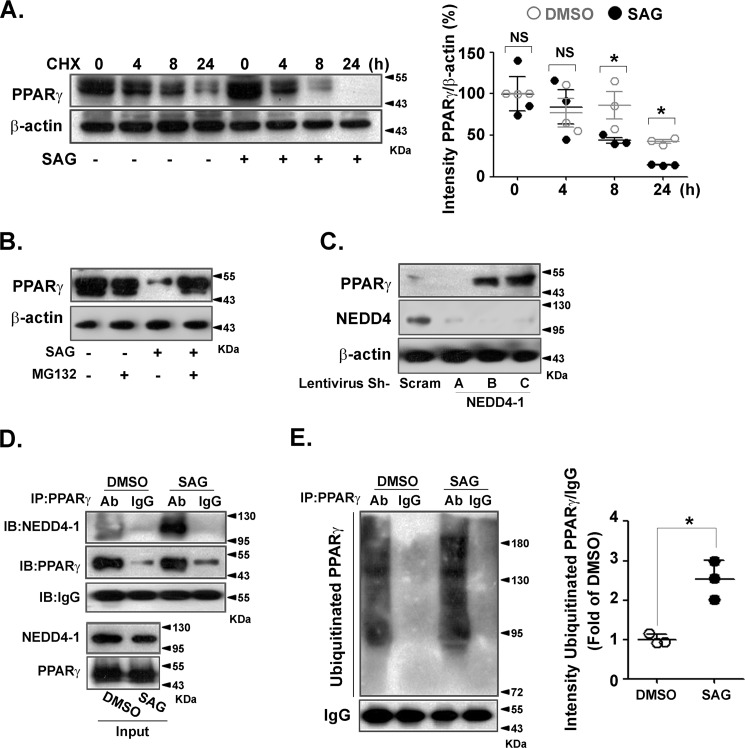
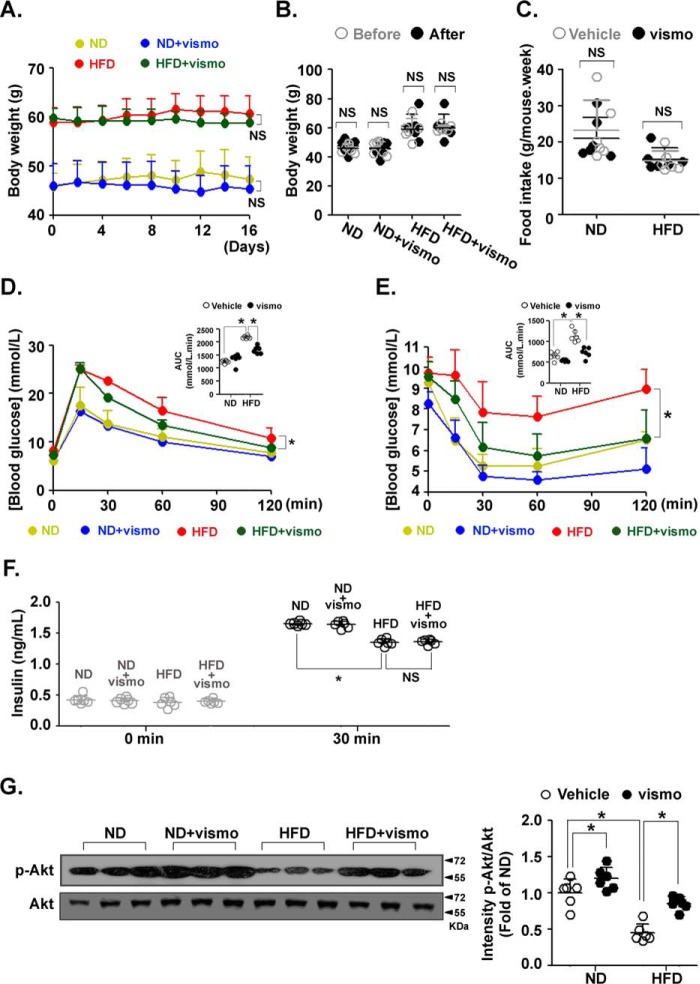
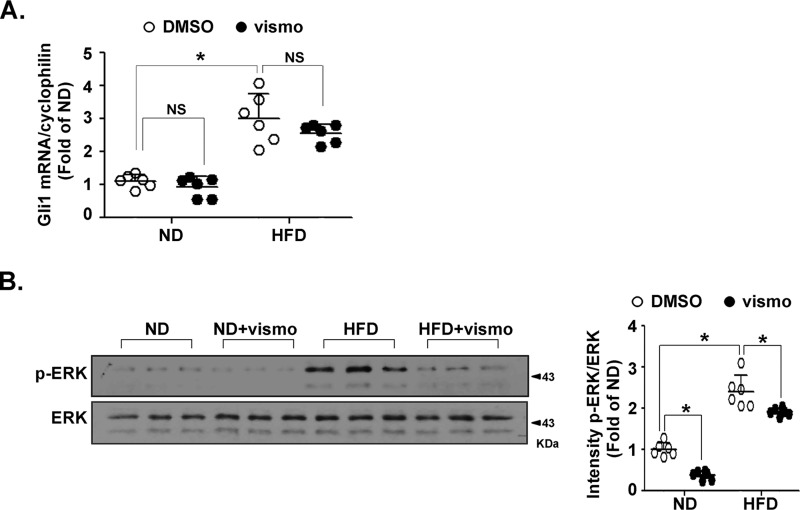
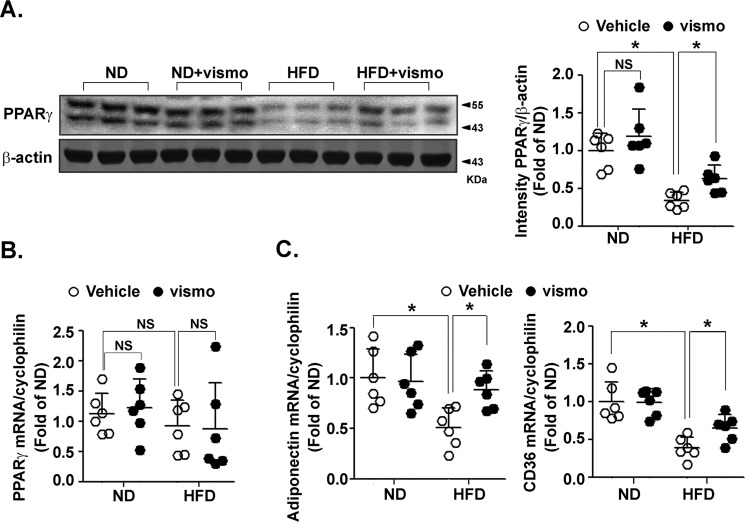
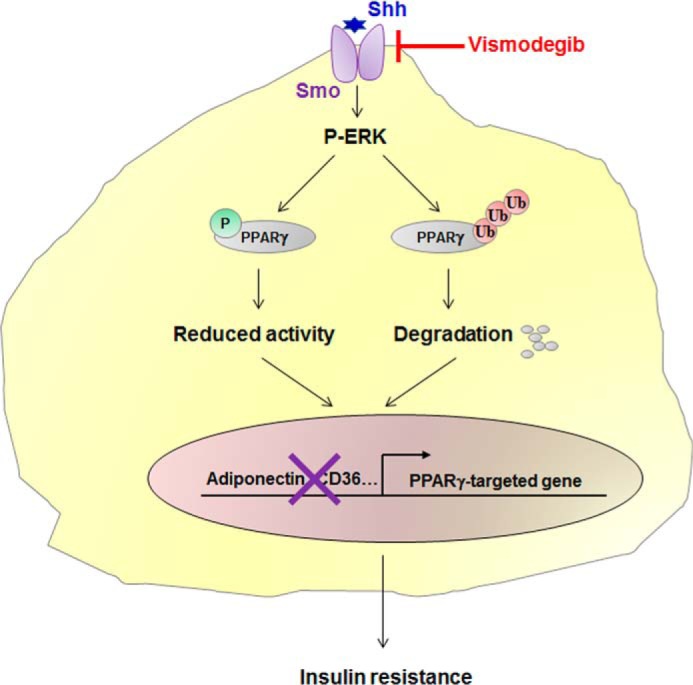
Obesity is a major risk for patients with chronic metabolic disorders including type 2 diabetes. Sonic hedgehog (Shh) is a morphogen that regulates the pancreas and adipose tissue formation during embryonic development. Peroxisome proliferator-activated receptor γ (PPARγ) is a member of the nuclear receptor superfamily and one of the most important regulators of insulin action. Here, we evaluated the role and mechanism of Shh signaling in obesity-associated insulin resistance and characterized its effect on PPARγ. We showed that Shh expression was up-regulated in subcutaneous fat from obese mice. In differentiated 3T3-L1 and primary cultured adipocytes from rats, recombinant Shh protein and SAG (an agonist of Shh signaling) activated an extracellular signal-regulated kinase (ERK)-dependent noncanonical pathway and induced PPARγ phosphorylation at serine 112, which decreased PPARγ activity. Meanwhile, Shh signaling degraded PPARγ protein via binding of PPARγ to neural precursor cell-expressed developmentally down-regulated protein 4-1 (NEDD4-1). Furthermore, vismodegib, an inhibitor of Shh signaling, attenuated ERK phosphorylation induced by a high fat diet (HFD) and restored PPARγ protein level, thus ameliorating glucose intolerance and insulin resistance in obese mice. Our finding suggests that Shh in subcutaneous fat decreases PPARγ activity and stability via activation of an ERK-dependent noncanonical pathway, resulting in impaired insulin action. Inhibition of Shh may serve as a potential therapeutic approach to treat obesity-related diabetes.
Keywords: NEDD4-1; insulin resistance; obesity; peroxisome proliferator-activated receptor (PPAR); protein degradation; sonic hedgehog (SHH); type 2 diabetes; ubiquitylation (ubiquitination).
© 2019 Yao et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
