

Inhibition of checkpoint kinase 1 following gemcitabine-mediated S phase arrest results in CDC7- and CDK2-dependent replication catastrophe
- PMID: 30573684
- PMCID: PMC6369309
- DOI: 10.1074/jbc.RA118.005231
Inhibition of checkpoint kinase 1 following gemcitabine-mediated S phase arrest results in CDC7- and CDK2-dependent replication catastrophe
Abstract
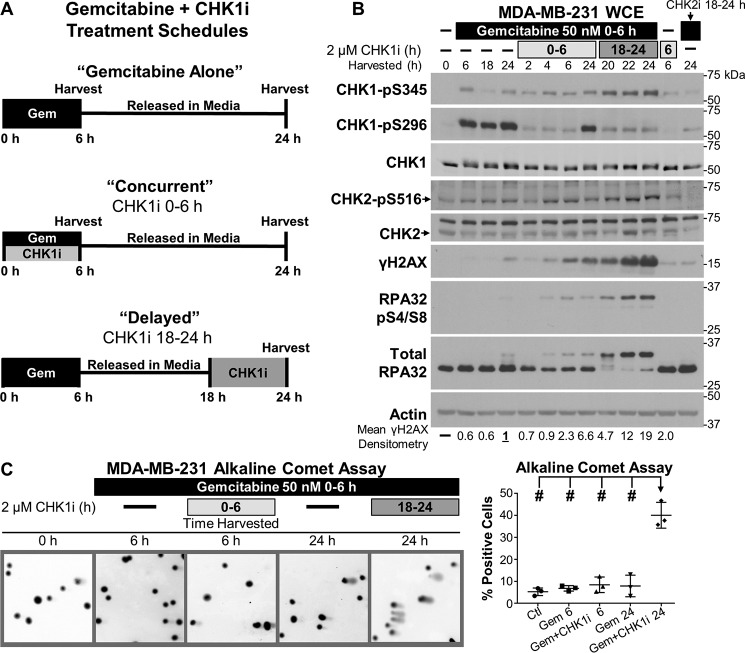
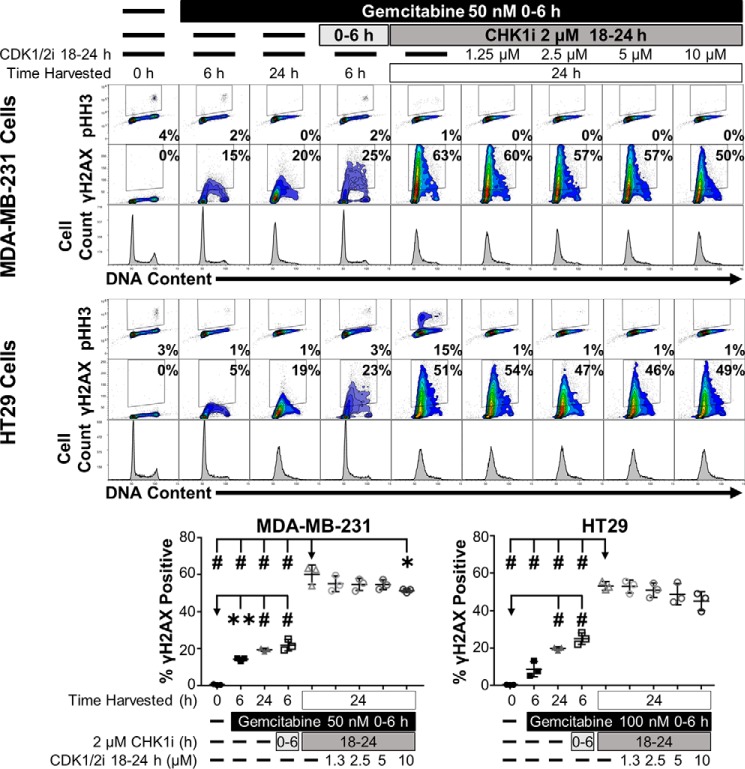
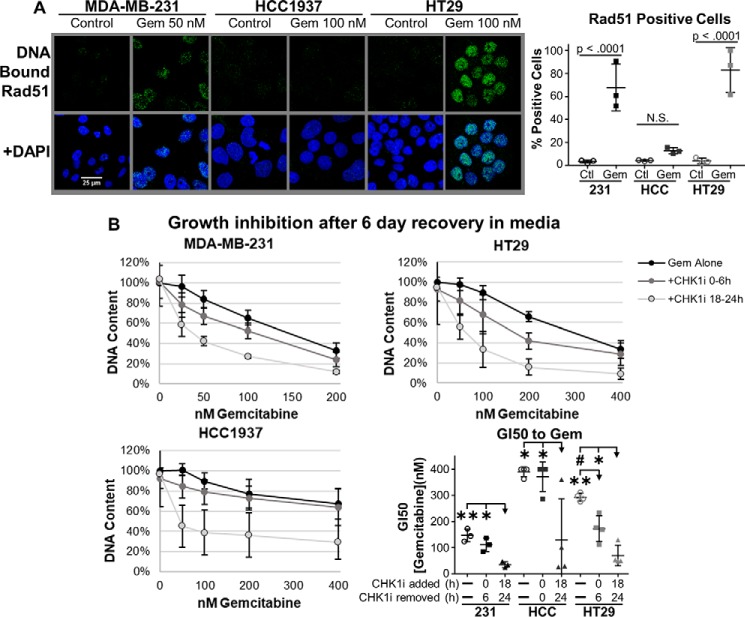
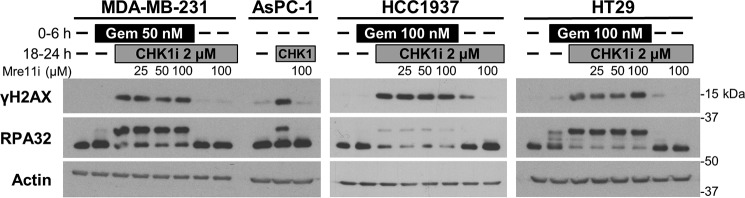
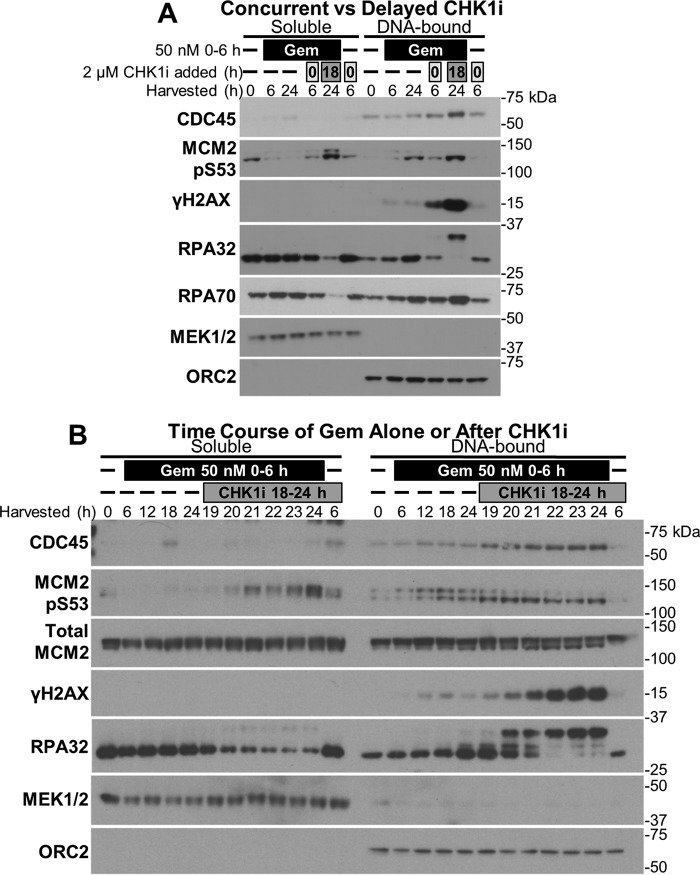
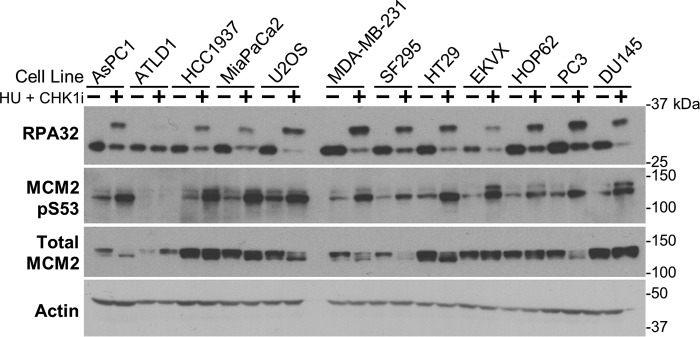
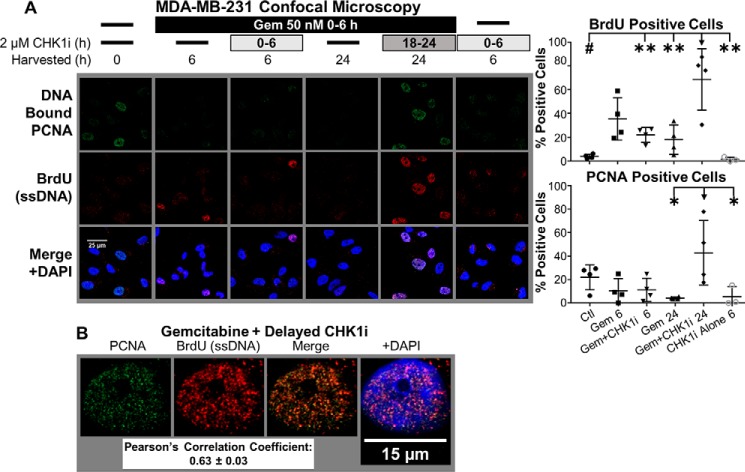
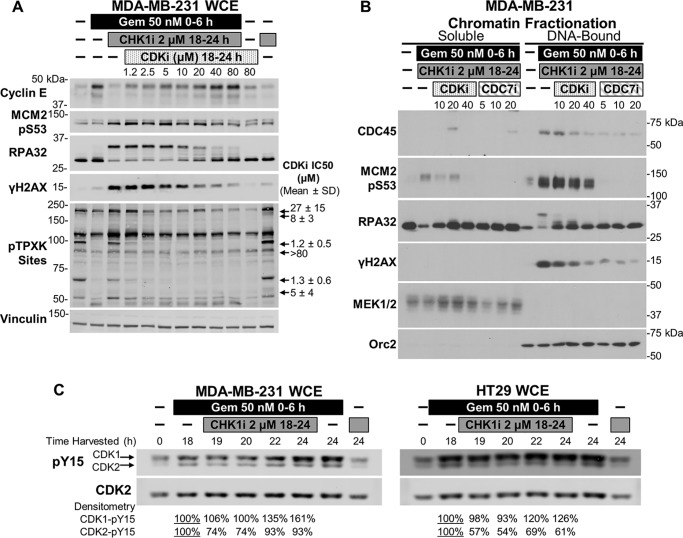
Combining DNA-damaging drugs with DNA checkpoint inhibitors is an emerging strategy to manage cancer. Checkpoint kinase 1 inhibitors (CHK1is) sensitize most cancer cell lines to DNA-damaging drugs and also elicit single-agent cytotoxicity in 15% of cell lines. Consequently, combination therapy may be effective in a broader patient population. Here, we characterized the molecular mechanism of sensitization to gemcitabine by the CHK1i MK8776. Brief gemcitabine incubation irreversibly inhibited ribonucleotide reductase, depleting dNTPs, resulting in durable S phase arrest. Addition of CHK1i 18 h after gemcitabine elicited cell division cycle 7 (CDC7)- and cyclin-dependent kinase 2 (CDK2)-dependent reactivation of the replicative helicase, but did not reinitiate DNA synthesis due to continued lack of dNTPs. Helicase reactivation generated extensive single-strand (ss)DNA that exceeded the protective capacity of the ssDNA-binding protein, replication protein A. The subsequent cleavage of unprotected ssDNA has been termed replication catastrophe. This mechanism did not occur with concurrent CHK1i plus gemcitabine treatment, providing support for delayed administration of CHK1i in patients. Alternative mechanisms of CHK1i-mediated sensitization to gemcitabine have been proposed, but their role was ruled out; these mechanisms include premature mitosis, inhibition of homologous recombination, and activation of double-strand break repair nuclease (MRE11). In contrast, single-agent activity of CHK1i was MRE11-dependent and was prevented by lower concentrations of a CDK2 inhibitor. Hence, both pathways require CDK2 but appear to depend on different CDK2 substrates. We conclude that a small-molecule inhibitor of CHK1 can elicit at least two distinct, context-dependent mechanisms of cytotoxicity in cancer cells.
Keywords: Chk1; DNA-damage response; cancer; cell cycle; cell division cycle 7-related protein kinase (Cdc7); cyclin-dependent kinase (CDK); gemcitabine; replication catastrophe; single-strand binding protein RPA.
© 2019 Warren and Eastman.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
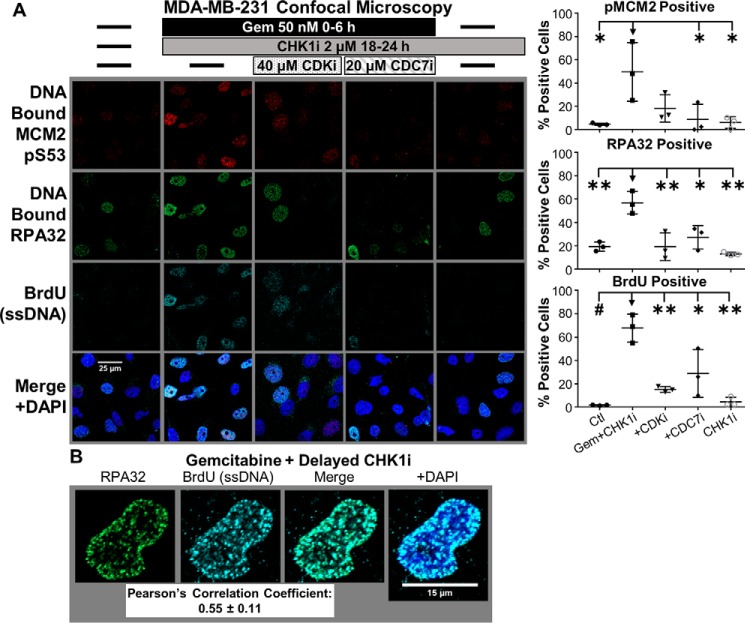
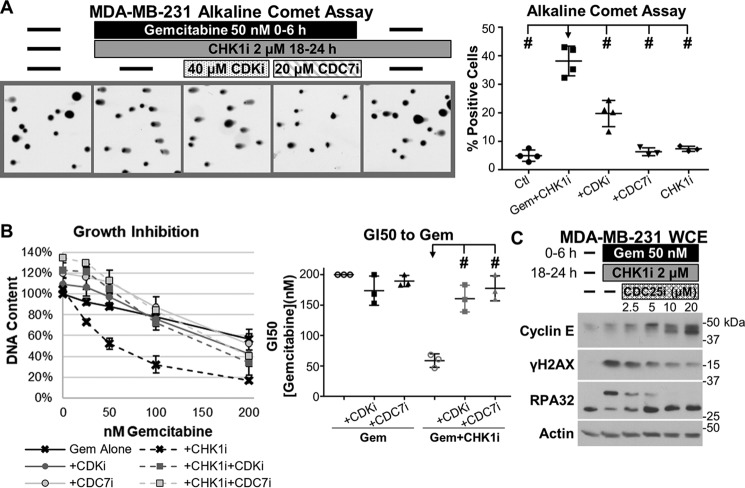
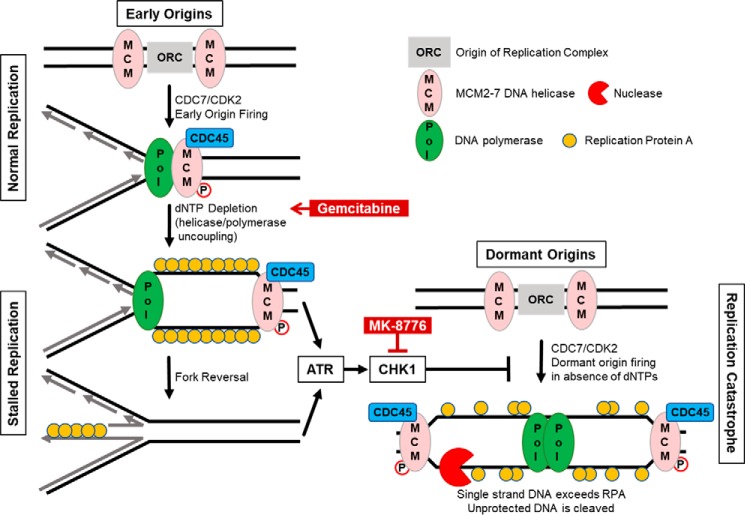
Comment in
-
Signaling dynamics of DNA damage response invoked by combination therapy are dose-dependent.J Biol Chem. 2019 Feb 8;294(6):2191. doi: 10.1074/jbc.L119.007381. J Biol Chem. 2019. PMID: 30737318 Free PMC article. No abstract available.
Similar articles
-
Comparison of the different mechanisms of cytotoxicity induced by checkpoint kinase I inhibitors when used as single agents or in combination with DNA damage.Oncogene. 2020 Feb;39(7):1389-1401. doi: 10.1038/s41388-019-1079-9. Epub 2019 Oct 28. Oncogene. 2020. PMID: 31659257 Free PMC article. Review.
-
A subset of cancer cell lines is acutely sensitive to the Chk1 inhibitor MK-8776 as monotherapy due to CDK2 activation in S phase.Oncotarget. 2016 Jan 12;7(2):1380-94. doi: 10.18632/oncotarget.6364. Oncotarget. 2016. PMID: 26595527 Free PMC article.
-
The contribution of DNA replication stress marked by high-intensity, pan-nuclear γH2AX staining to chemosensitization by CHK1 and WEE1 inhibitors.Cell Cycle. 2018;17(9):1076-1086. doi: 10.1080/15384101.2018.1475827. Epub 2018 Jul 18. Cell Cycle. 2018. PMID: 29895190 Free PMC article.
-
Chk1 inhibition after replicative stress activates a double strand break response mediated by ATM and DNA-dependent protein kinase.Cell Cycle. 2010 Mar 1;9(5):995-1004. doi: 10.4161/cc.9.5.10935. Epub 2010 Mar 14. Cell Cycle. 2010. PMID: 20160494
-
Cyclin-dependent kinases and S phase control in mammalian cells.Cell Cycle. 2003 Jul-Aug;2(4):316-24. Cell Cycle. 2003. PMID: 12851482 Review.
Cited by
-
Ribonucleotide Reductases: Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets.Annu Rev Biochem. 2020 Jun 20;89:45-75. doi: 10.1146/annurev-biochem-013118-111843. Annu Rev Biochem. 2020. PMID: 32569524 Free PMC article. Review.
-
Everything in Moderation: Lessons Learned by Exploiting Moderate Replication Stress in Cancer.Cancers (Basel). 2019 Sep 6;11(9):1320. doi: 10.3390/cancers11091320. Cancers (Basel). 2019. PMID: 31500184 Free PMC article. Review.
-
Comparison of the different mechanisms of cytotoxicity induced by checkpoint kinase I inhibitors when used as single agents or in combination with DNA damage.Oncogene. 2020 Feb;39(7):1389-1401. doi: 10.1038/s41388-019-1079-9. Epub 2019 Oct 28. Oncogene. 2020. PMID: 31659257 Free PMC article. Review.
-
DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer.Signal Transduct Target Ther. 2020 May 1;5(1):60. doi: 10.1038/s41392-020-0150-x. Signal Transduct Target Ther. 2020. PMID: 32355263 Free PMC article. Review.
-
Inhibition of Protein Synthesis Induced by CHK1 Inhibitors Discriminates Sensitive from Resistant Cancer Cells.ACS Pharmacol Transl Sci. 2021 Aug 4;4(4):1449-1461. doi: 10.1021/acsptsci.1c00150. eCollection 2021 Aug 13. ACS Pharmacol Transl Sci. 2021. PMID: 34423276 Free PMC article.
References
-
- Brown J. S., O'Carrigan B., Jackson S. P., and Yap T. A. (2017) Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 7, 20–37 10.1158/2159-8290.CD-16-0860 - DOI - PMC - PubMed
-
- Bunch R. T., and Eastman A. (1996) Enhancement of cisplatin-induced cytotoxicity by 7-hydroxystaurosporine (UCN-01) a new G2-checkpoint inhibitor. Clin. Cancer Res. 2, 791–797 - PubMed
-
- Montano R., Thompson R., Chung I., Hou H., Khan N., and Eastman A. (2013) Sensitization of human cancer cells to gemcitabine by the Chk1 inhibitor MK-8776: cell cycle perturbation and impact of administration of schedule in vitro and in vivo. BMC Cancer 13, 604 10.1186/1471-2407-13-604 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
