

Estrogen receptor α promotes protein synthesis by fine-tuning the expression of the eukaryotic translation initiation factor 3 subunit f (eIF3f)
- PMID: 30573685
- PMCID: PMC6378971
- DOI: 10.1074/jbc.RA118.004383
Estrogen receptor α promotes protein synthesis by fine-tuning the expression of the eukaryotic translation initiation factor 3 subunit f (eIF3f)
Abstract
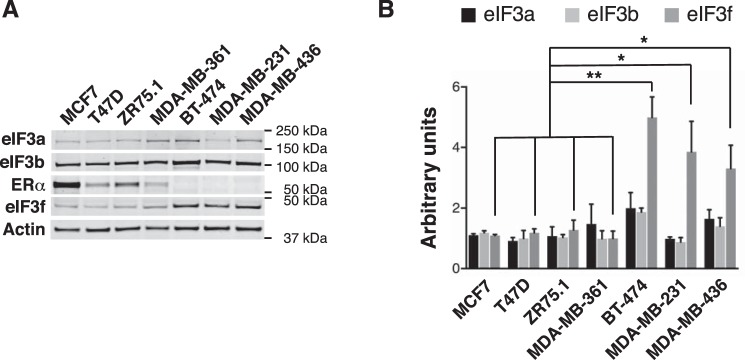
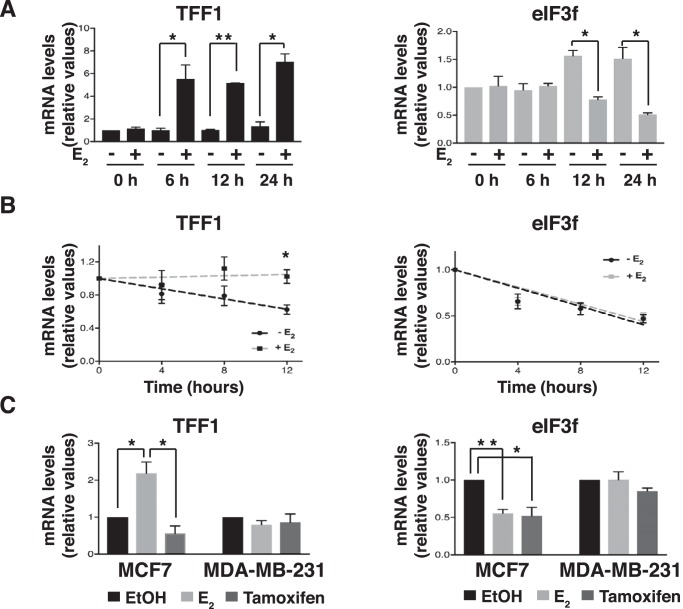
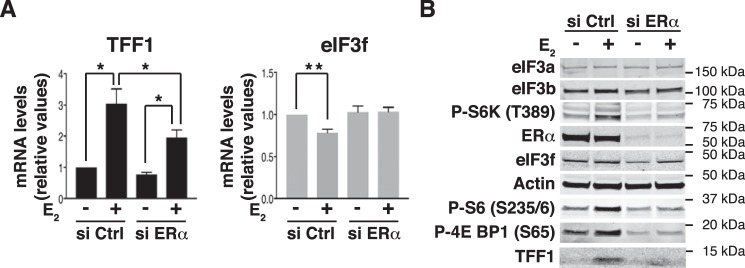
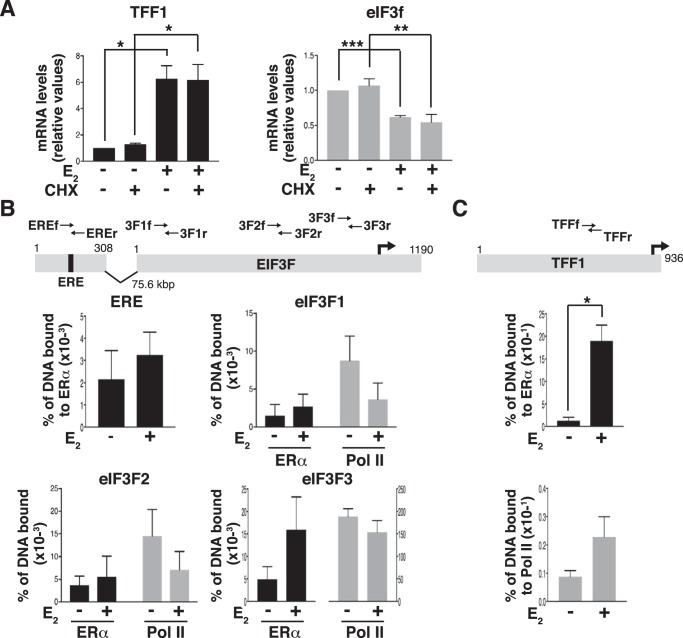
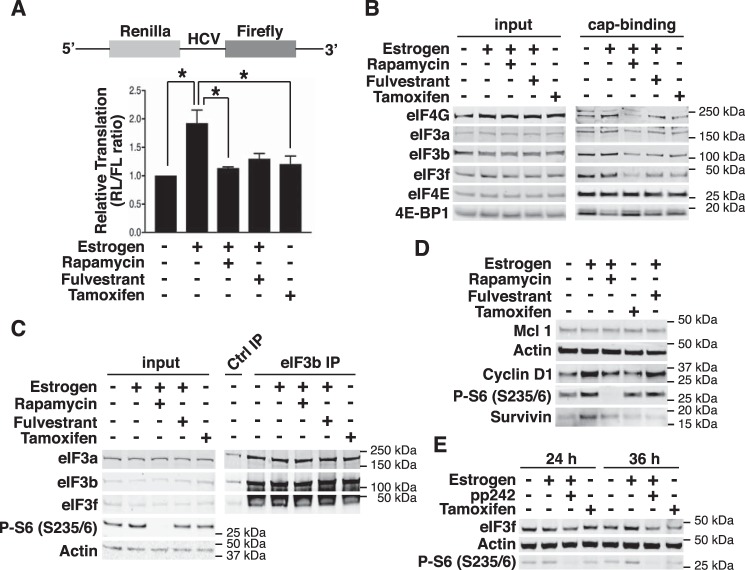
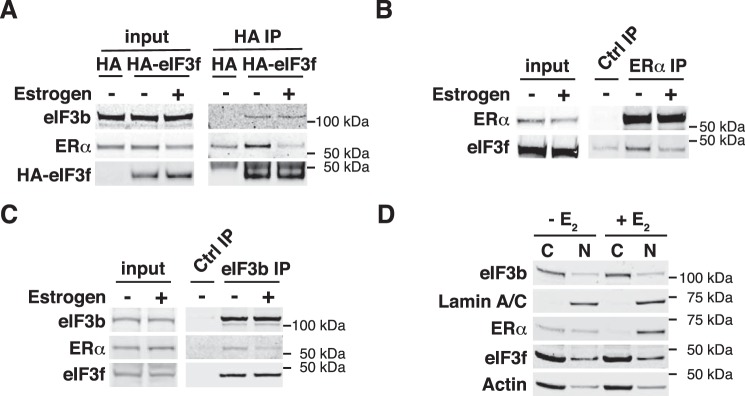
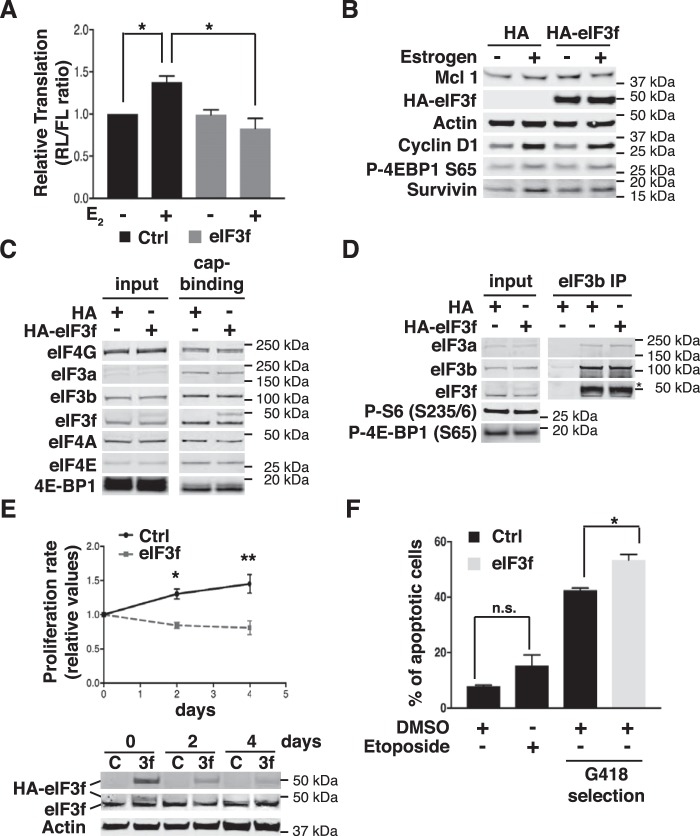
Approximately two thirds of all breast cancer cases are estrogen receptor (ER)-positive. The treatment of this breast cancer subtype with endocrine therapies is effective in the adjuvant and recurrent settings. However, their effectiveness is compromised by the emergence of intrinsic or acquired resistance. Thus, identification of new molecular targets can significantly contribute to the development of novel therapeutic strategies. In recent years, many studies have implicated aberrant levels of translation initiation factors in cancer etiology and provided evidence that identifies these factors as promising therapeutic targets. Accordingly, we observed reduced levels of the eIF3 subunit eIF3f in ER-positive breast cancer cells compared with ER-negative cells, and determined that low eIF3f levels are required for proper proliferation and survival of ER-positive MCF7 cells. The expression of eIF3f is tightly controlled by ERα at the transcriptional (genomic pathway) and translational (nongenomic pathway) level. Specifically, estrogen-bound ERα represses transcription of the EIF3F gene, while promoting eIF3f mRNA translation. To regulate translation, estrogen activates the mTORC1 pathway, which enhances the binding of eIF3 to the eIF4F complex and, consequently, the assembly of the 48S preinitiation complexes and protein synthesis. We observed preferential translation of mRNAs with highly structured 5'-UTRs that usually encode factors involved in cell proliferation and survival (e.g. cyclin D1 and survivin). Our results underscore the importance of estrogen-ERα-mediated control of eIF3f expression for the proliferation and survival of ER-positive breast cancer cells. These findings may provide rationale for the development of new therapies to treat ER-positive breast cancer.
Keywords: ERα; breast cancer; eIF3f; estrogen receptor; eukaryotic translation initiation; gene expression; mTORC1; translation.
© 2019 Cuesta et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Carroll J. S., Meyer C. A., Song J., Li W., Geistlinger T. R., Eeckhoute J., Brodsky A. S., Keeton E. K., Fertuck K. C., Hall G. F., Wang Q., Bekiranov S., Sementchenko V., Fox E. A., Silver P. A., Gingeras T. R., Liu X. S., and Brown M. (2006) Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 38, 1289–1297 10.1038/ng1901 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
