

Calmodulinopathy: A Novel, Life-Threatening Clinical Entity Affecting the Young
- PMID: 30574507
- PMCID: PMC6291462
- DOI: 10.3389/fcvm.2018.00175
Calmodulinopathy: A Novel, Life-Threatening Clinical Entity Affecting the Young
Abstract
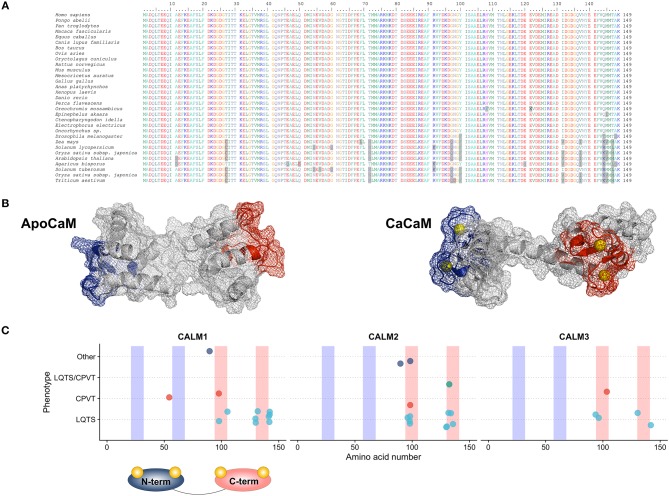
Sudden cardiac death (SCD) in the young may often be the first manifestation of a genetic arrythmogenic disease that had remained undiagnosed. Despite the significant discoveries of the genetic bases of inherited arrhythmia syndromes, there remains a measurable fraction of cases where in-depth clinical and genetic investigations fail to identify the underlying SCD etiology. A few years ago, 2 cases of infants with recurrent cardiac arrest episodes, due to what appeared to be as a severe form of long QT syndrome (LQTS), came to our attention. These prompted a number of clinical and genetic research investigations that allowed us to identify a novel, closely associated to LQTS but nevertheless distinct, clinical entity that is now known as calmodulinopathy. Calmodulinopathy is a life-threatening arrhythmia syndrome, affecting mostly young individuals, caused by mutations in any of the 3 genes encoding calmodulin (CaM). Calmodulin is a ubiquitously expressed Ca2+ signaling protein that, in the heart, modulates several ion channels and participates in a plethora of cellular processes. We will hereby provide an overview of CaM's structure and function under normal and disease states, highlighting the genetic etiology of calmodulinopathy and the related disease mechanisms. We will also discuss the phenotypic spectrum of patients with calmodulinopathy and present state-of-the art approaches with patient-derived induced pluripotent stem cells that have been thus far adopted in order to accurately model calmodulinopathy in vitro, decipher disease mechanisms and identify novel therapies.
Keywords: CALM; calmodulin; catecholaminergic polymorphic ventricular tachycardia; long QT syndrome; sudden cardiac death.
Figures


Similar articles
-
Calmodulinopathy in inherited arrhythmia syndromes.Tzu Chi Med J. 2021 Apr 14;33(4):339-344. doi: 10.4103/tcmj.tcmj_182_20. eCollection 2021 Oct-Dec. Tzu Chi Med J. 2021. PMID: 34760628 Free PMC article. Review.
-
Calmodulin mutations and life-threatening cardiac arrhythmias: insights from the International Calmodulinopathy Registry.Eur Heart J. 2019 Sep 14;40(35):2964-2975. doi: 10.1093/eurheartj/ehz311. Eur Heart J. 2019. PMID: 31170290 Free PMC article.
-
Clinical presentation of calmodulin mutations: the International Calmodulinopathy Registry.Eur Heart J. 2023 Sep 14;44(35):3357-3370. doi: 10.1093/eurheartj/ehad418. Eur Heart J. 2023. PMID: 37528649 Free PMC article.
-
A Precision Medicine Approach to the Rescue of Function on Malignant Calmodulinopathic Long-QT Syndrome.Circ Res. 2017 Jan 6;120(1):39-48. doi: 10.1161/CIRCRESAHA.116.309283. Epub 2016 Oct 20. Circ Res. 2017. PMID: 27765793 Free PMC article.
-
Calmodulinopathies: throwing back the veil on the newest life-threatening genetic arrhythmia syndrome.Curr Opin Cardiol. 2021 Jan;36(1):61-66. doi: 10.1097/HCO.0000000000000808. Curr Opin Cardiol. 2021. PMID: 33027101 Review.
Cited by
-
Calmodulinopathy: Functional Effects of CALM Mutations and Their Relationship With Clinical Phenotypes.Front Cardiovasc Med. 2018 Dec 11;5:176. doi: 10.3389/fcvm.2018.00176. eCollection 2018. Front Cardiovasc Med. 2018. PMID: 30619883 Free PMC article. Review.
-
The Prenatal Diagnosis and Perinatal Management of Congenital Long QT Syndrome: A Comprehensive Literature Review and Recent Updates.J Cardiovasc Dev Dis. 2025 Apr 14;12(4):156. doi: 10.3390/jcdd12040156. J Cardiovasc Dev Dis. 2025. PMID: 40278215 Free PMC article. Review.
-
The mechanism of LQTS related CaM mutation E141G interfering with CaV1.2 channels function through its C-lobe.J Physiol Biochem. 2025 Feb;81(1):185-197. doi: 10.1007/s13105-024-01064-5. Epub 2024 Dec 19. J Physiol Biochem. 2025. PMID: 39699847
-
Gene therapy for cardiac arrhythmias.Nat Rev Cardiol. 2025 May 23. doi: 10.1038/s41569-025-01168-5. Online ahead of print. Nat Rev Cardiol. 2025. PMID: 40410593 Review.
-
Calmodulin mutation in long QT syndrome 15 associated with congenital heart defects further complicated by a functional 2:1 atrioventricular block: Management from foetal life to postpartum.Indian Pacing Electrophysiol J. 2024 May-Jun;24(3):150-154. doi: 10.1016/j.ipej.2024.01.006. Epub 2024 Jan 27. Indian Pacing Electrophysiol J. 2024. PMID: 38281621 Free PMC article.
References
-
- Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. . ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC)endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. (2015) 36:2793–867. 10.1093/eurheartj/ehv316 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous