

DDX58 and Classic Singleton-Merten Syndrome
- PMID: 30574673
- PMCID: PMC6394545
- DOI: 10.1007/s10875-018-0572-1
DDX58 and Classic Singleton-Merten Syndrome
Abstract
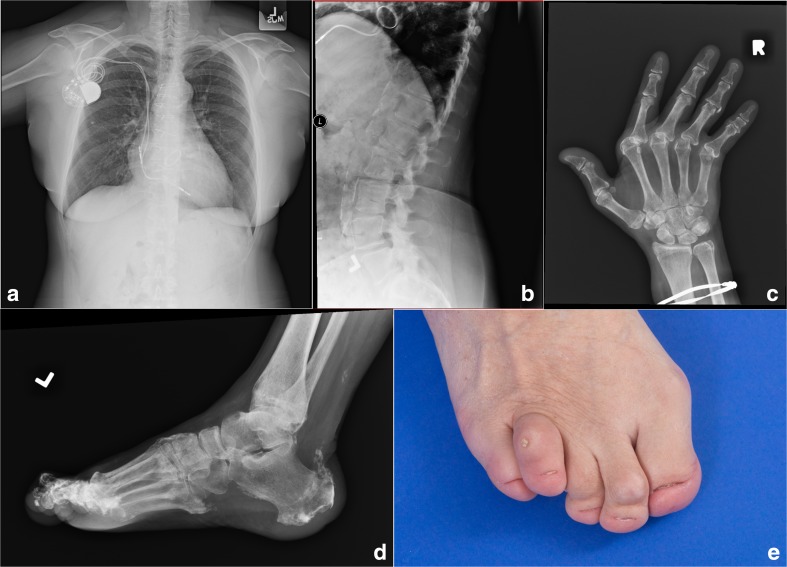
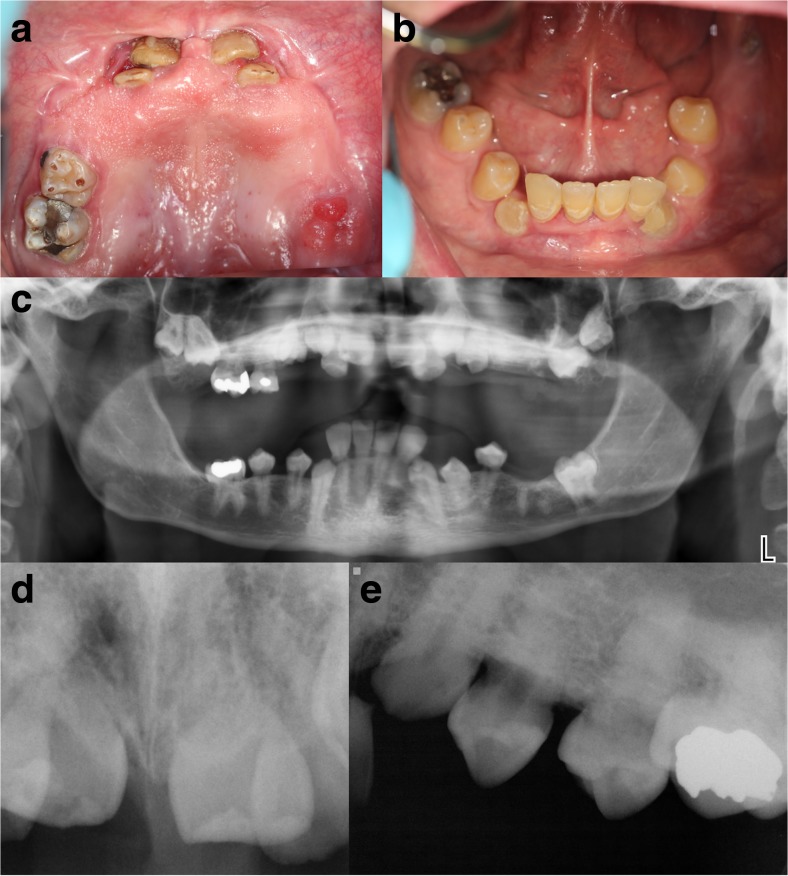
Purpose: Singleton-Merten syndrome manifests as dental dysplasia, glaucoma, psoriasis, aortic calcification, and skeletal abnormalities including tendon rupture and arthropathy. Pathogenic variants in IFIH1 have previously been associated with the classic Singleton-Merten syndrome, while variants in DDX58 has been described in association with a milder phenotype, which is suggested to have a better prognosis. We studied a family with severe, "classic" Singleton-Merten syndrome.
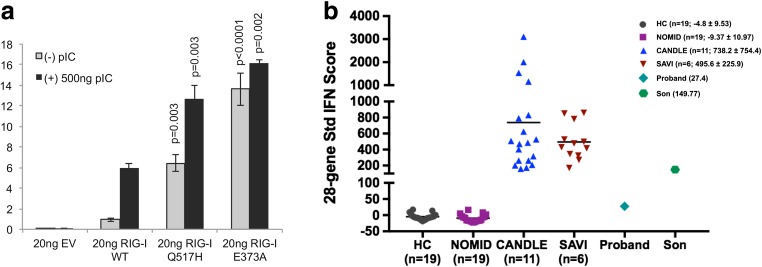
Methods: We undertook clinical phenotyping, next-generation sequencing, and functional studies of type I interferon production in patient whole blood and assessed the type I interferon promoter activity in HEK293 cells transfected with wild-type or mutant DDX58 stimulated with Poly I:C.
Results: We demonstrate a DDX58 autosomal dominant gain-of-function mutation, with constitutive upregulation of type I interferon.
Conclusions: DDX58 mutations may be associated with the classic features of Singleton-Merten syndrome including dental dysplasia, tendon rupture, and severe cardiac sequela.
Keywords: Interferonopathy; Singleton-Merten syndrome; retinoic acid-inducible gene I; type I interferon.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Toward a better understanding of type I interferonopathies: a brief summary, update and beyond.World J Pediatr. 2020 Feb;16(1):44-51. doi: 10.1007/s12519-019-00273-z. Epub 2019 Aug 3. World J Pediatr. 2020. PMID: 31377974 Review.
-
Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome.Am J Hum Genet. 2015 Feb 5;96(2):266-74. doi: 10.1016/j.ajhg.2014.11.019. Epub 2015 Jan 22. Am J Hum Genet. 2015. PMID: 25620203 Free PMC article.
-
Musculoskeletal Disease in MDA5-Related Type I Interferonopathy: A Mendelian Mimic of Jaccoud's Arthropathy.Arthritis Rheumatol. 2017 Oct;69(10):2081-2091. doi: 10.1002/art.40179. Epub 2017 Aug 22. Arthritis Rheumatol. 2017. PMID: 28605144 Free PMC article.
-
Further evidence for specific IFIH1 mutation as a cause of Singleton-Merten syndrome with phenotypic heterogeneity.Am J Med Genet A. 2017 May;173(5):1396-1399. doi: 10.1002/ajmg.a.38214. Epub 2017 Mar 20. Am J Med Genet A. 2017. PMID: 28319323
-
MDA5-Associated Neuroinflammation and the Singleton-Merten Syndrome: Two Faces of the Same Type I Interferonopathy Spectrum.J Interferon Cytokine Res. 2017 May;37(5):214-219. doi: 10.1089/jir.2017.0004. J Interferon Cytokine Res. 2017. PMID: 28475458 Free PMC article. Review.
Cited by
-
Genetic and epigenetic factors shape phenotypes and outcomes in systemic lupus erythematosus - focus on juvenile-onset systemic lupus erythematosus.Curr Opin Rheumatol. 2025 Mar 1;37(2):149-163. doi: 10.1097/BOR.0000000000001072. Epub 2024 Dec 11. Curr Opin Rheumatol. 2025. PMID: 39660463 Free PMC article. Review.
-
Toward a better understanding of type I interferonopathies: a brief summary, update and beyond.World J Pediatr. 2020 Feb;16(1):44-51. doi: 10.1007/s12519-019-00273-z. Epub 2019 Aug 3. World J Pediatr. 2020. PMID: 31377974 Review.
-
Established and Emerging Roles of DEAD/H-Box Helicases in Regulating Infection and Immunity.Immunol Rev. 2025 Jan;329(1):e13426. doi: 10.1111/imr.13426. Epub 2024 Dec 2. Immunol Rev. 2025. PMID: 39620586 Free PMC article. Review.
-
Dermatologic and Dermatopathologic Features of Monogenic Autoinflammatory Diseases.Front Immunol. 2019 Oct 29;10:2448. doi: 10.3389/fimmu.2019.02448. eCollection 2019. Front Immunol. 2019. PMID: 31736939 Free PMC article. Review.
-
The molecular language of RNA 5' ends: guardians of RNA identity and immunity.RNA. 2024 Mar 18;30(4):327-336. doi: 10.1261/rna.079942.124. RNA. 2024. PMID: 38325897 Free PMC article. Review.
References
-
- Rutsch F, MacDougall M, Lu C, Buers I, Mamaeva O, Nitschke Y, Rice GI, Erlandsen H, Kehl HG, Thiele H, Nürnberg P, Höhne W, Crow YJ, Feigenbaum A, Hennekam RC. A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am J Hum Genet. 2015;96(2):275–282. doi: 10.1016/j.ajhg.2014.12.014. - DOI - PMC - PubMed
-
- Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, Nishikomori R, Funatsuka M, Ohshima Y, Sugawara Y, Yasumi T, Kato H, Shirai T, Ohara O, Fujita T, Heike T. Aicardi-Goutières syndrome is caused by IFIH1 mutations. Am J Hum Genet. 2014;95(1):121–125. doi: 10.1016/j.ajhg.2014.06.007. - DOI - PMC - PubMed
-
- de Carvalho LM, Ngoumou G, Park JW, Ehmke N, Deigendesch N, Kitabayashi N, Melki I, Souza FFL, Tzschach A, Nogueira-Barbosa MH, Ferriani V, Louzada-Junior P, Marques W, Jr, Lourenço CM, Horn D, Kallinich T, Stenzel W, Hur S, Rice GI, Crow YJ. Musculoskeletal disease in MDA5-related type I interferonopathy: a Mendelian mimic of Jaccoud’s arthropathy. Arthritis Rheumatol. 2017;69(10):2081–2091. doi: 10.1002/art.40179. - DOI - PMC - PubMed
-
- Bursztejn AC, Briggs TA, del Toro Duany Y, Anderson BH, O’Sullivan J, Williams SG, Bodemer C, Fraitag S, Gebhard F, Leheup B, Lemelle I, Oojageer A, Raffo E, Schmitt E, Rice GI, Hur S, Crow YJ. Unusual cutaneous features associated with a heterozygous gain-of-function mutation in IFIH1: overlap between Aicardi-Goutières and Singleton-Merten syndromes. Br J Dermatol. 2015;173(6):1505–1513. doi: 10.1111/bjd.14073. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
