

Sorafenib for Advanced and Refractory Desmoid Tumors
- PMID: 30575484
- PMCID: PMC6447029
- DOI: 10.1056/NEJMoa1805052
Sorafenib for Advanced and Refractory Desmoid Tumors
Abstract
Background: Desmoid tumors (also referred to as aggressive fibromatosis) are connective tissue neoplasms that can arise in any anatomical location and infiltrate the mesentery, neurovascular structures, and visceral organs. There is no standard of care.
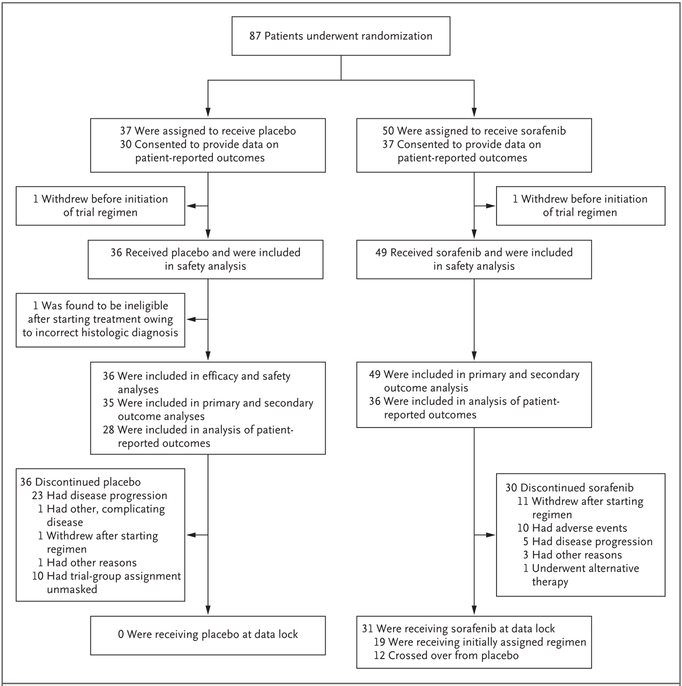
Methods: In this double-blind, phase 3 trial, we randomly assigned 87 patients with progressive, symptomatic, or recurrent desmoid tumors to receive either sorafenib (400-mg tablet once daily) or matching placebo. Crossover to the sorafenib group was permitted for patients in the placebo group who had disease progression. The primary end point was investigator-assessed progression-free survival; rates of objective response and adverse events were also evaluated.
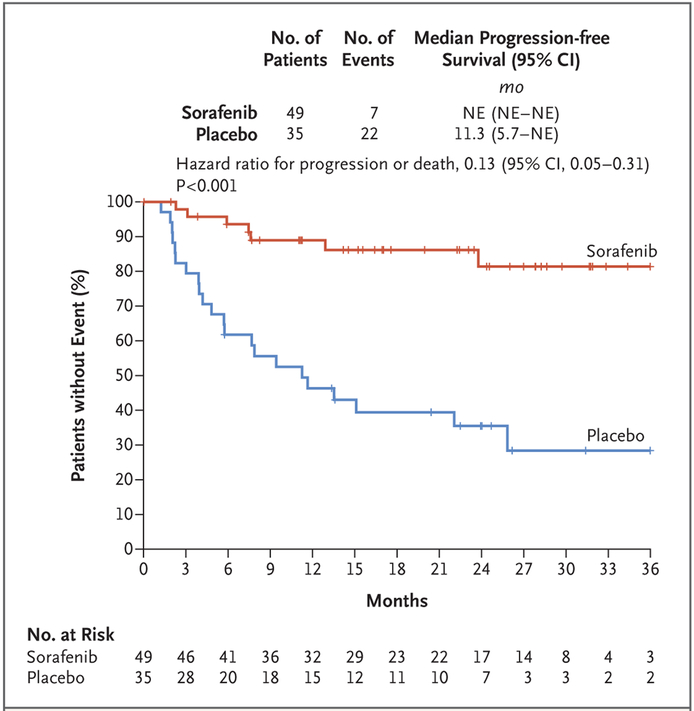
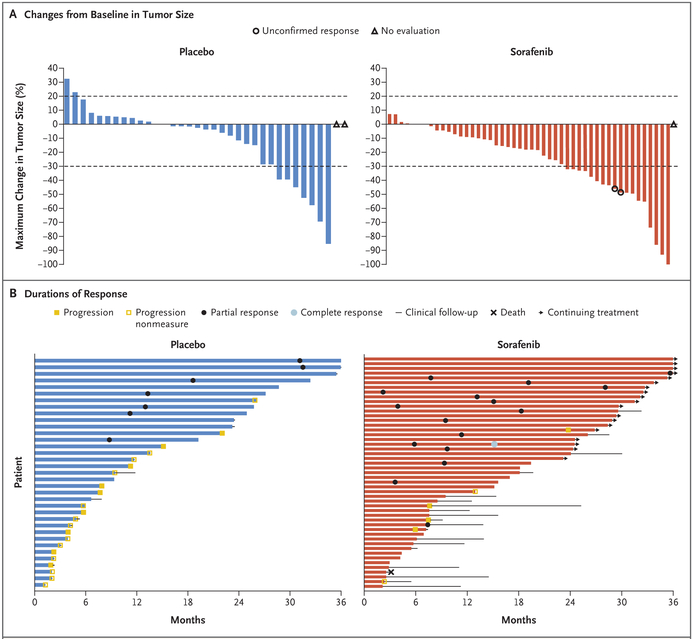
Results: With a median follow-up of 27.2 months, the 2-year progression-free survival rate was 81% (95% confidence interval [CI], 69 to 96) in the sorafenib group and 36% (95% CI, 22 to 57) in the placebo group (hazard ratio for progression or death, 0.13; 95% CI, 0.05 to 0.31; P<0.001). Before crossover, the objective response rate was 33% (95% CI, 20 to 48) in the sorafenib group and 20% (95% CI, 8 to 38) in the placebo group. The median time to an objective response among patients who had a response was 9.6 months (interquartile range, 6.6 to 16.7) in the sorafenib group and 13.3 months (interquartile range, 11.2 to 31.1) in the placebo group. The objective responses are ongoing. Among patients who received sorafenib, the most frequently reported adverse events were grade 1 or 2 events of rash (73%), fatigue (67%), hypertension (55%), and diarrhea (51%).
Conclusions: Among patients with progressive, refractory, or symptomatic desmoid tumors, sorafenib significantly prolonged progression-free survival and induced durable responses. (Funded by the National Cancer Institute and others; ClinicalTrials.gov number, NCT02066181 .).
Figures
Comment in
-
Targeting PCSK9 to reduce residual risk in ACS.Nat Rev Cardiol. 2019 Jan;16(1):2. doi: 10.1038/s41569-018-0125-6. Nat Rev Cardiol. 2019. PMID: 30478399 No abstract available.
-
Desmoid Tumors Respond to Sorafenib.Cancer Discov. 2019 Mar;9(3):313. doi: 10.1158/2159-8290.CD-NB2019-004. Epub 2019 Jan 22. Cancer Discov. 2019. PMID: 30670460
References
-
- Fletcher CDM, Bridge JA, Hogendoorn P, Mertens F, eds. WHO classification of tumours of soft tissue and bone. 4th ed. Lyon, France: IARC Press, 2013.
-
- Brennan MF, Antonescu CR, Alektiar K, Maki RG. Management of soft tissue sarcoma. Cham, Switzerland: Springer, 2016.
-
- Salas S, Chibon F, Noguchi T, et al. Molecular characterization by array comparative genomic hybridization and DNA sequencing of 194 desmoid tumors. Genes Chromosomes Cancer 2010;49:560–8. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- UG1 CA233329/CA/NCI NIH HHS/United States
- U10 CA180858/CA/NCI NIH HHS/United States
- UG1 CA189850/CA/NCI NIH HHS/United States
- U10 CA180863/CA/NCI NIH HHS/United States
- U24 CA196171/CA/NCI NIH HHS/United States
- U10 CA180888/CA/NCI NIH HHS/United States
- U10 CA180868/CA/NCI NIH HHS/United States
- U10 CA180826/CA/NCI NIH HHS/United States
- P30 CA086862/CA/NCI NIH HHS/United States
- U10 CA180821/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- U10 CA180857/CA/NCI NIH HHS/United States
- U10 CA180882/CA/NCI NIH HHS/United States
- R01 FD005105/FD/FDA HHS/United States
- U10 CA180820/CA/NCI NIH HHS/United States
- U10 CA180833/CA/NCI NIH HHS/United States
- UG1 CA189957/CA/NCI NIH HHS/United States
- U10 CA180838/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical