

Human genome-wide measurement of drug-responsive regulatory activity
- PMID: 30575722
- PMCID: PMC6303339
- DOI: 10.1038/s41467-018-07607-x
Human genome-wide measurement of drug-responsive regulatory activity
Abstract
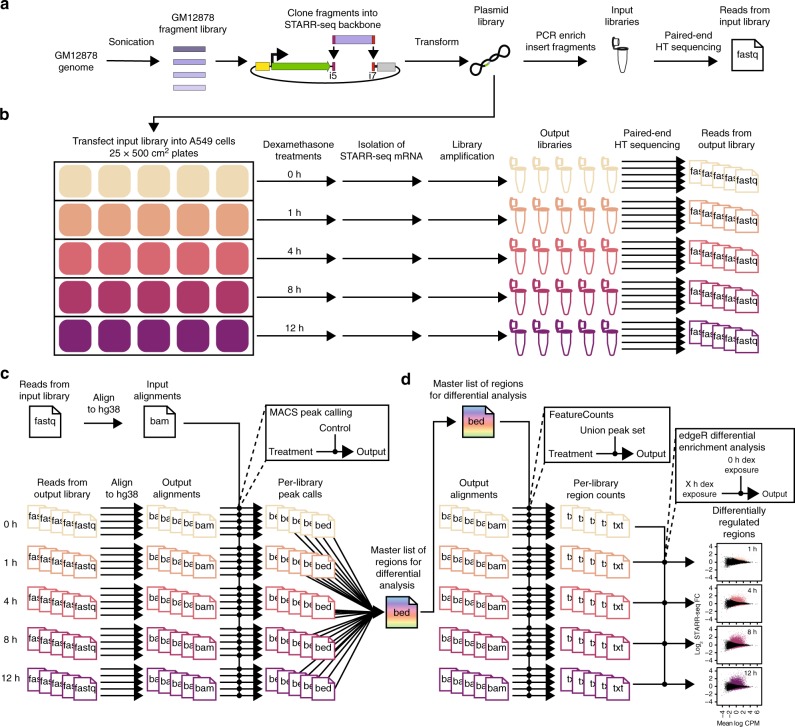
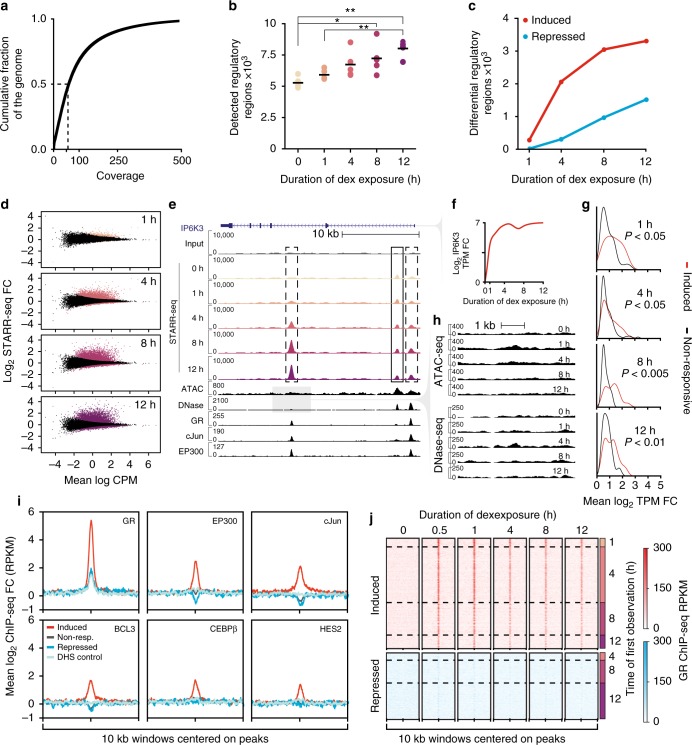
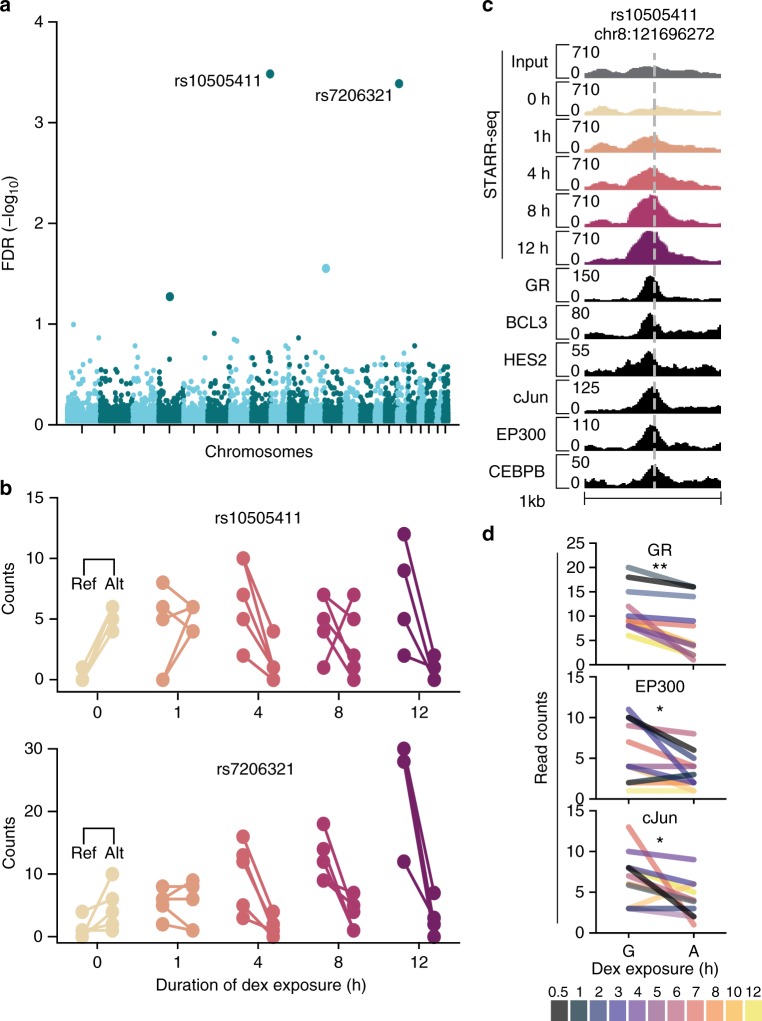
Environmental stimuli commonly act via changes in gene regulation. Human-genome-scale assays to measure such responses are indirect or require knowledge of the transcription factors (TFs) involved. Here, we present the use of human genome-wide high-throughput reporter assays to measure environmentally-responsive regulatory element activity. We focus on responses to glucocorticoids (GCs), an important class of pharmaceuticals and a paradigmatic genomic response model. We assay GC-responsive regulatory activity across >108 unique DNA fragments, covering the human genome at >50×. Those assays directly detected thousands of GC-responsive regulatory elements genome-wide. We then validate those findings with measurements of transcription factor occupancy, histone modifications, chromatin accessibility, and gene expression. We also detect allele-specific environmental responses. Notably, the assays did not require knowledge of GC response mechanisms. Thus, this technology can be used to agnostically quantify genomic responses for which the underlying mechanism remains unknown.
Conflict of interest statement
T.E.R. is a co-founder and consultant for Element Genomics which uses high-throughput reporter assays to study gene regulation. The remaining authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
