

Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells
- PMID: 30575730
- PMCID: PMC6303303
- DOI: 10.1038/s41467-018-07824-4
Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells
Erratum in
-
Author Correction: Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells.Nat Commun. 2022 Apr 1;13(1):1894. doi: 10.1038/s41467-022-29467-2. Nat Commun. 2022. PMID: 35365656 Free PMC article. No abstract available.
Abstract
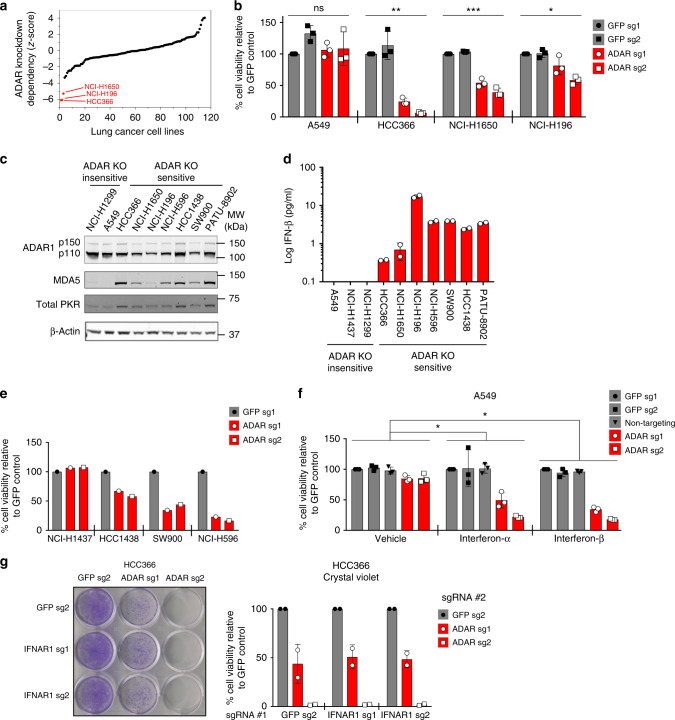
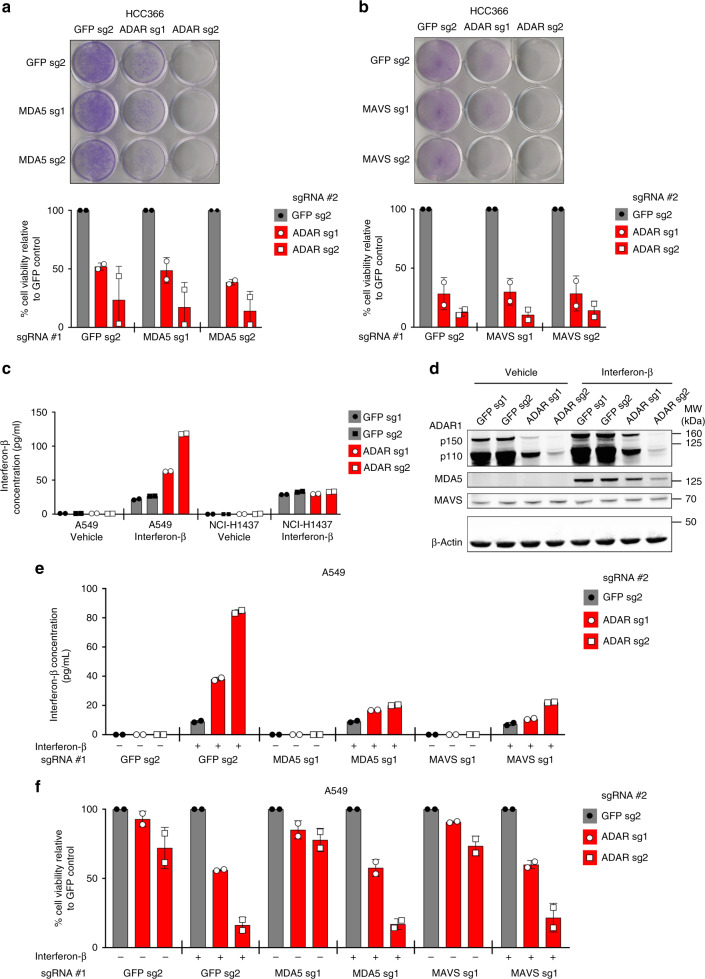
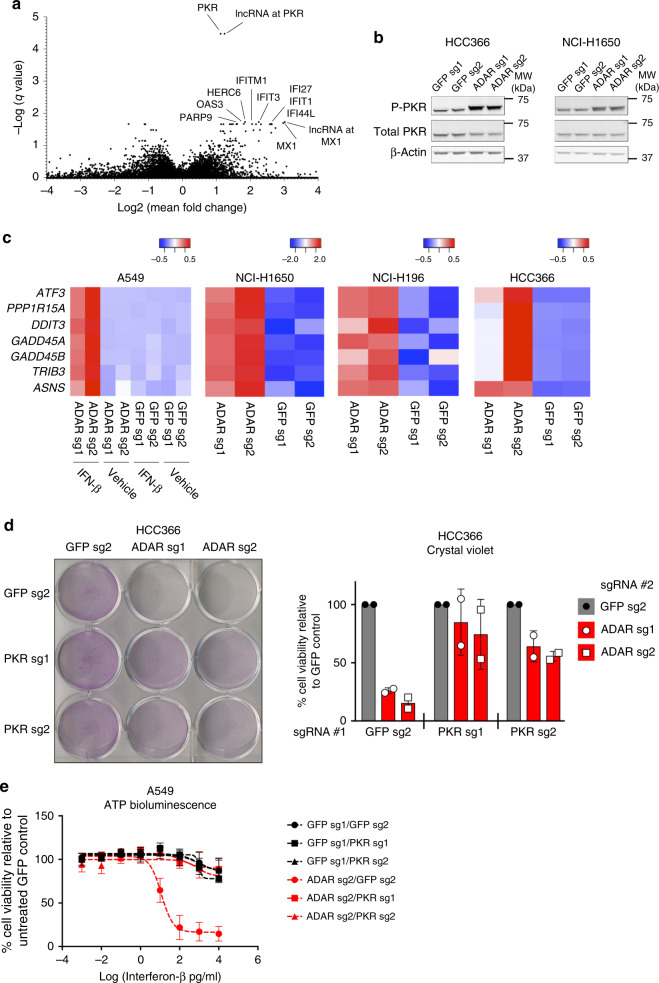
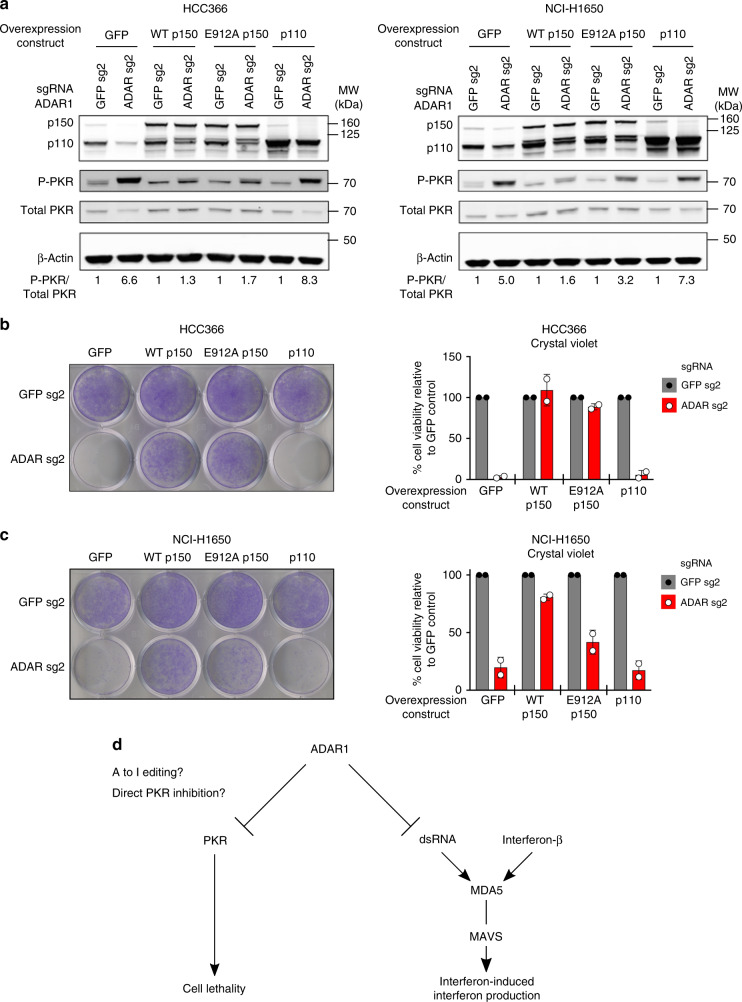
Systematic exploration of cancer cell vulnerabilities can inform the development of novel cancer therapeutics. Here, through analysis of genome-scale loss-of-function datasets, we identify adenosine deaminase acting on RNA (ADAR or ADAR1) as an essential gene for the survival of a subset of cancer cell lines. ADAR1-dependent cell lines display increased expression of interferon-stimulated genes. Activation of type I interferon signaling in the context of ADAR1 deficiency can induce cell lethality in non-ADAR1-dependent cell lines. ADAR deletion causes activation of the double-stranded RNA sensor, protein kinase R (PKR). Disruption of PKR signaling, through inactivation of PKR or overexpression of either a wildtype or catalytically inactive mutant version of the p150 isoform of ADAR1, partially rescues cell lethality after ADAR1 loss, suggesting that both catalytic and non-enzymatic functions of ADAR1 may contribute to preventing PKR-mediated cell lethality. Together, these data nominate ADAR1 as a potential therapeutic target in a subset of cancers.
Conflict of interest statement
H.S.G. is now an employee of KSQ Therapeutics, Inc. in Cambridge, MA. W.C.H. is a founder of and consultant for KSQ Therapeutics, Inc. in Cambridge, MA. M.M. receives research funding from Bayer; had equity interest in Foundation Medicine (former, partially sold to Roche); had a consulting or advisory role with Foundation Medicine (former) and OrigiMed; and received patent royalties on intellectual property from Dana-Farber Cancer Institute licensed to LabCorp. M.M. and H.S.G. are inventors on a patent application for the use of ADAR1 inhibitors in cancer treatment. The remaining authors declare no competing interests.
Figures
References
-
- Paz-Ares, L. et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N. Engl. J. Med.10.1056/NEJMoa1810865 (2018). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
