

PARL deficiency in mouse causes Complex III defects, coenzyme Q depletion, and Leigh-like syndrome
- PMID: 30578322
- PMCID: PMC6320509
- DOI: 10.1073/pnas.1811938116
PARL deficiency in mouse causes Complex III defects, coenzyme Q depletion, and Leigh-like syndrome
Abstract
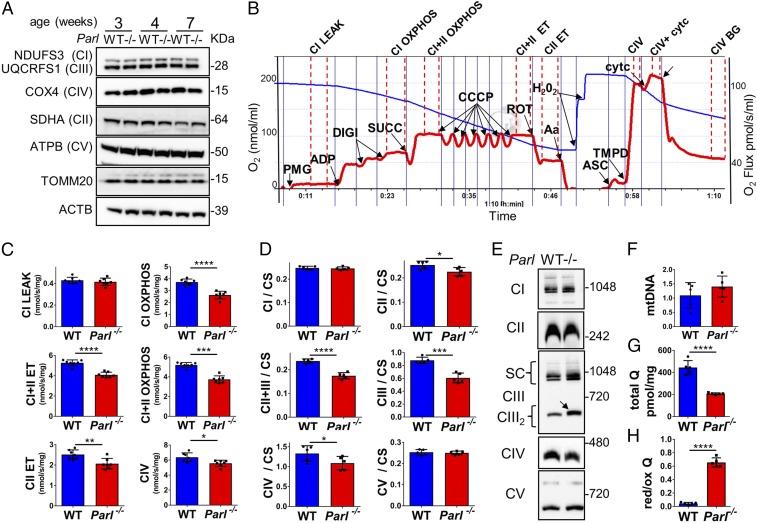
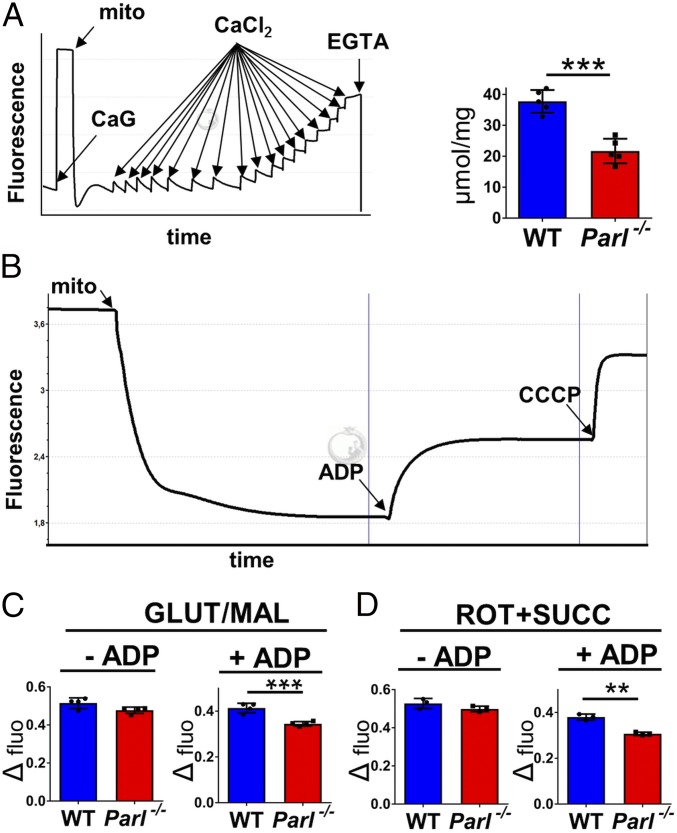
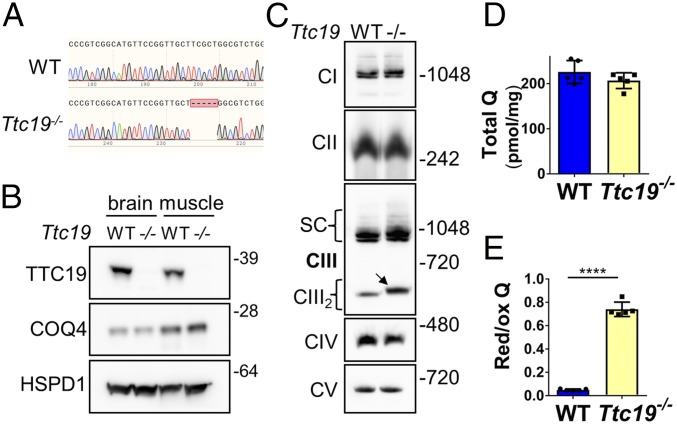
The mitochondrial intramembrane rhomboid protease PARL has been implicated in diverse functions in vitro, but its physiological role in vivo remains unclear. Here we show that Parl ablation in mouse causes a necrotizing encephalomyelopathy similar to Leigh syndrome, a mitochondrial disease characterized by disrupted energy production. Mice with conditional PARL deficiency in the nervous system, but not in muscle, develop a similar phenotype as germline Parl KOs, demonstrating the vital role of PARL in neurological homeostasis. Genetic modification of two major PARL substrates, PINK1 and PGAM5, do not modify this severe neurological phenotype. Parl-/- brain mitochondria are affected by progressive ultrastructural changes and by defects in Complex III (CIII) activity, coenzyme Q (CoQ) biosynthesis, and mitochondrial calcium metabolism. PARL is necessary for the stable expression of TTC19, which is required for CIII activity, and of COQ4, which is essential in CoQ biosynthesis. Thus, PARL plays a previously overlooked constitutive role in the maintenance of the respiratory chain in the nervous system, and its deficiency causes progressive mitochondrial dysfunction and structural abnormalities leading to neuronal necrosis and Leigh-like syndrome.
Keywords: Leigh syndrome; mitochondria; neurodegeneration; respiratory chain; rhomboid protease.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




References
-
- Spinazzi M, De Strooper B. PARL: The mitochondrial rhomboid protease. Semin Cell Dev Biol. 2016;60:19–28. - PubMed
-
- Düsterhöft S, Künzel U, Freeman M. Rhomboid proteases in human disease: Mechanisms and future prospects. Biochim Biophys Acta Mol Cell Res. 2017;1864:2200–2209. - PubMed
-
- Cipolat S, et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126:163–175. - PubMed
-
- Chao J-R, et al. Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature. 2008;452:98–102. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
