

Coupled Single-Cell CRISPR Screening and Epigenomic Profiling Reveals Causal Gene Regulatory Networks
- PMID: 30580963
- PMCID: PMC6329648
- DOI: 10.1016/j.cell.2018.11.022
Coupled Single-Cell CRISPR Screening and Epigenomic Profiling Reveals Causal Gene Regulatory Networks
Abstract
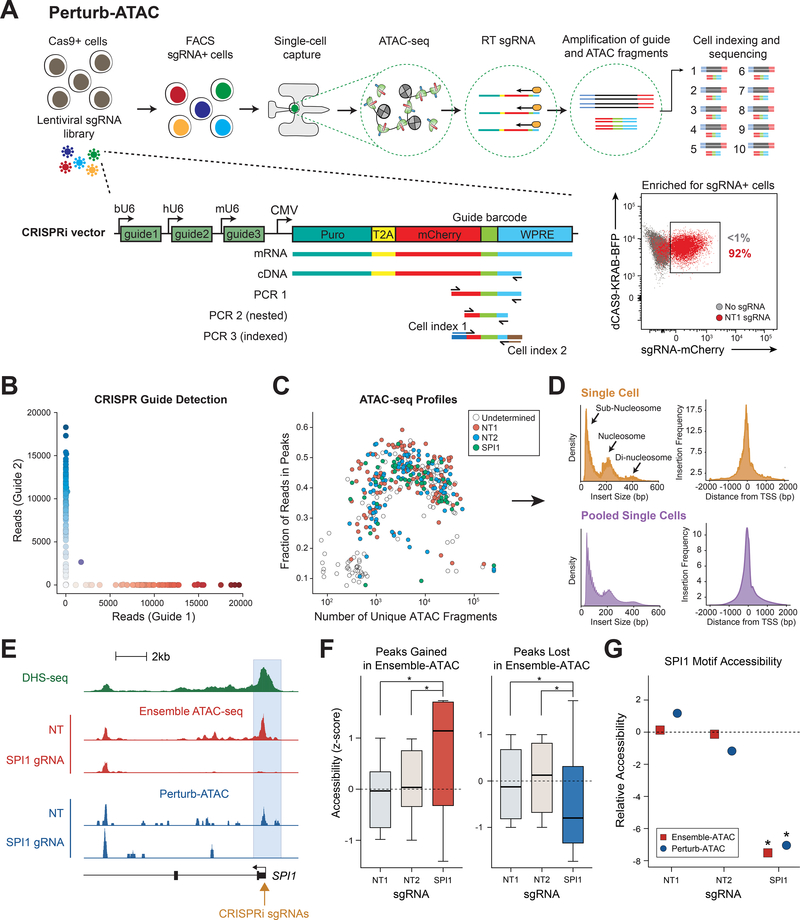
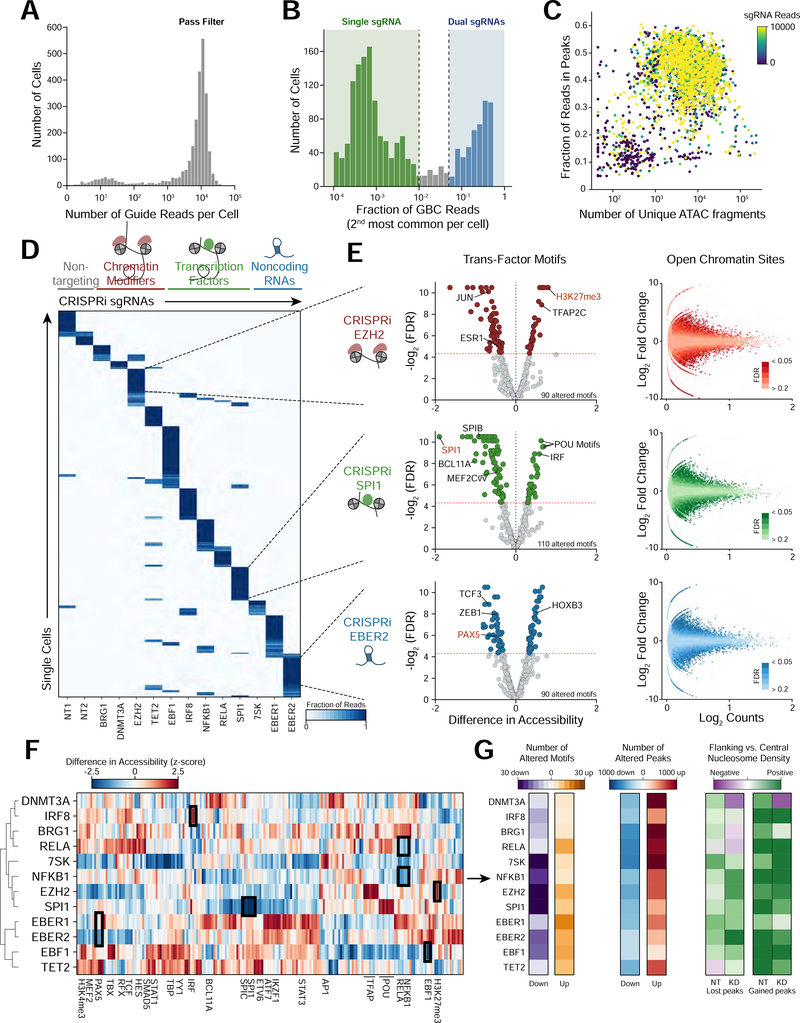
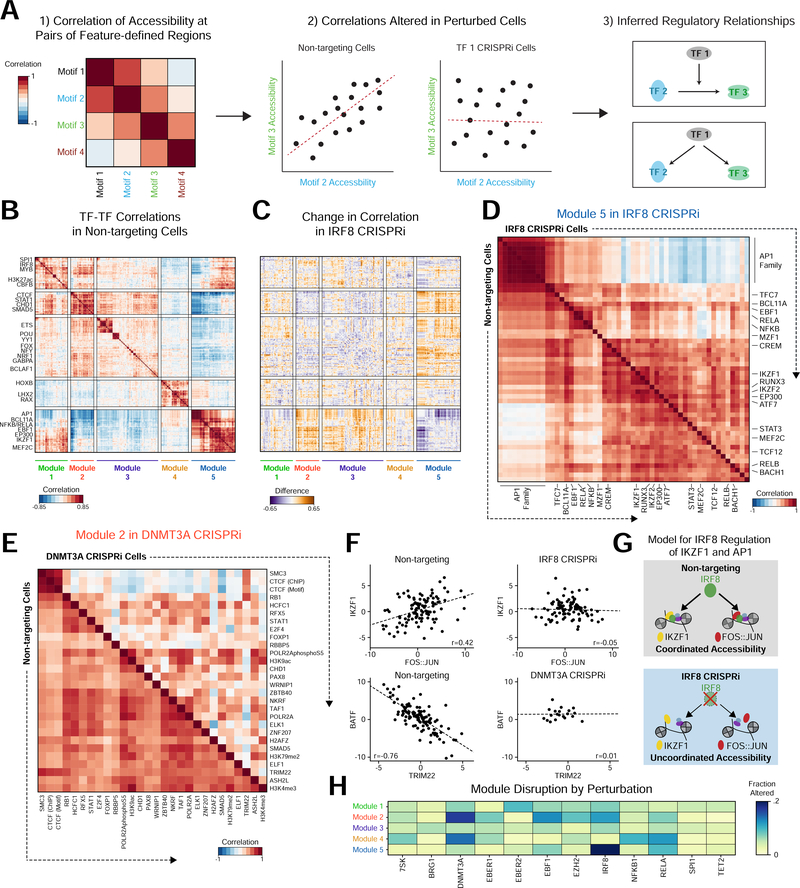
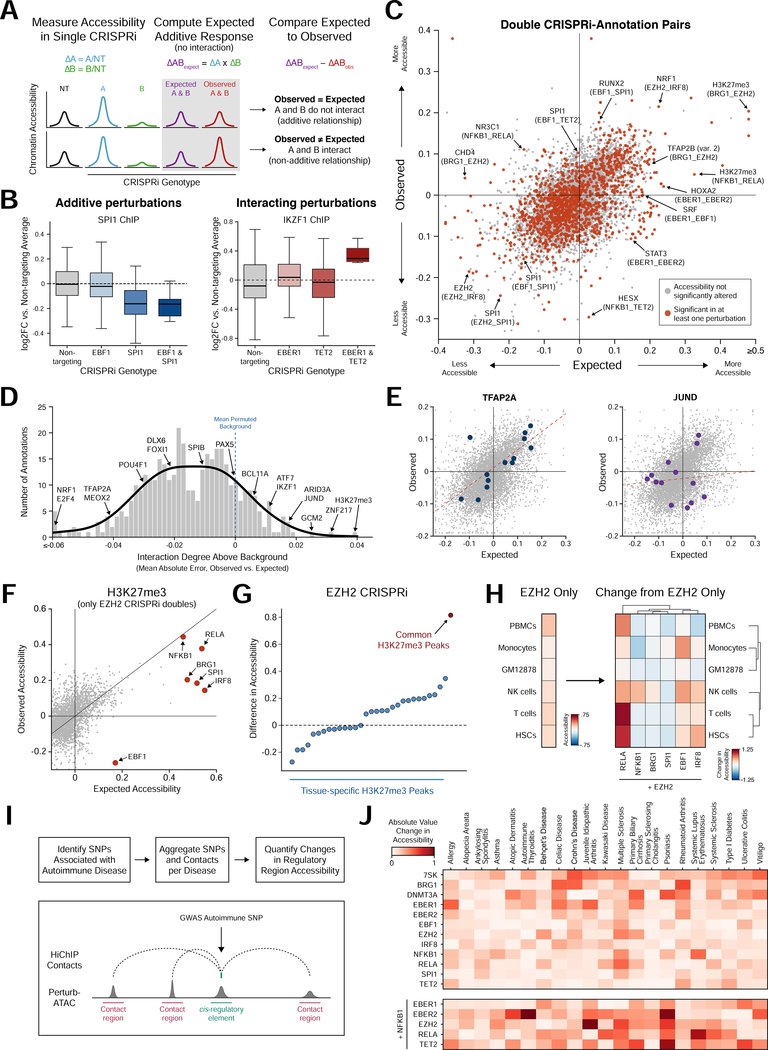
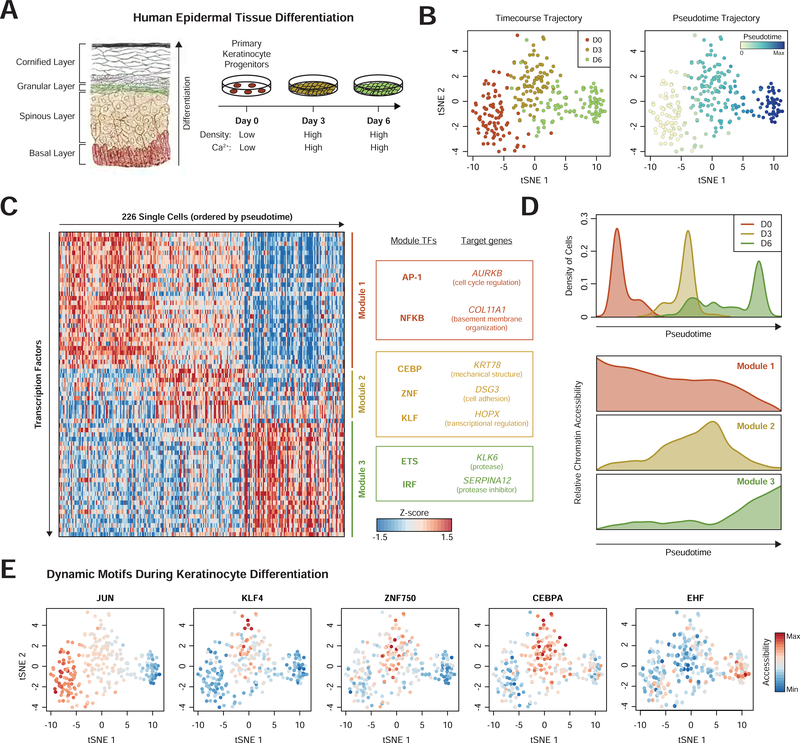
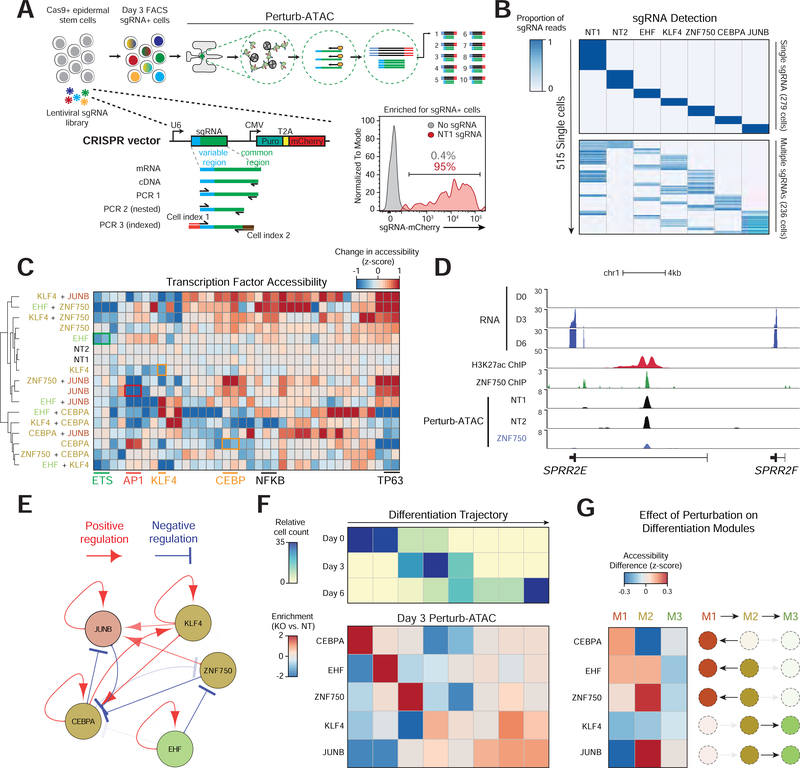
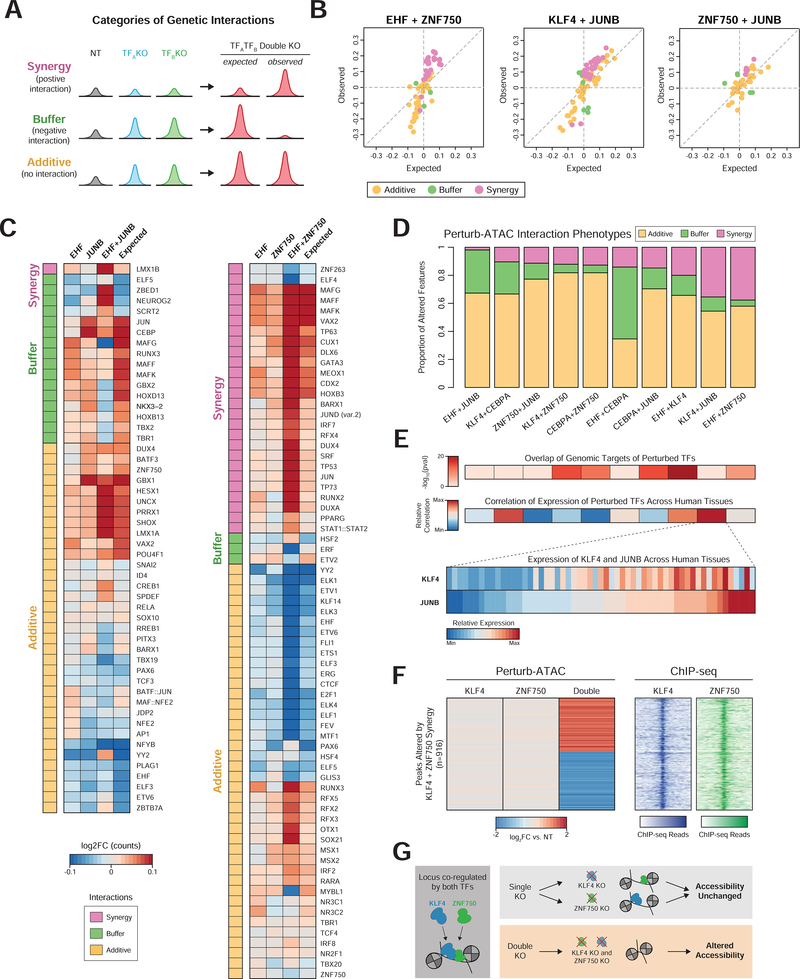
Here, we present Perturb-ATAC, a method that combines multiplexed CRISPR interference or knockout with genome-wide chromatin accessibility profiling in single cells based on the simultaneous detection of CRISPR guide RNAs and open chromatin sites by assay of transposase-accessible chromatin with sequencing (ATAC-seq). We applied Perturb-ATAC to transcription factors (TFs), chromatin-modifying factors, and noncoding RNAs (ncRNAs) in ∼4,300 single cells, encompassing more than 63 genotype-phenotype relationships. Perturb-ATAC in human B lymphocytes uncovered regulators of chromatin accessibility, TF occupancy, and nucleosome positioning and identified a hierarchy of TFs that govern B cell state, variation, and disease-associated cis-regulatory elements. Perturb-ATAC in primary human epidermal cells revealed three sequential modules of cis-elements that specify keratinocyte fate. Combinatorial deletion of all pairs of these TFs uncovered their epistatic relationships and highlighted genomic co-localization as a basis for synergistic interactions. Thus, Perturb-ATAC is a powerful strategy to dissect gene regulatory networks in development and disease.
Keywords: ATAC-seq; CRISPR; chromatin accessibility; epigenomics; pooled screens; single-cell genomics.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
References
-
- Adamson B, Norman TM, Jost M, and Weissman JS (2018). Approaches to maximize sgRNA-barcode coupling in Perturb-seq screens. BioRxiv 298349.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
