

Human Cellular Models for the Investigation of Lung Inflammation and Mucus Production in Cystic Fibrosis
- PMID: 30581723
- PMCID: PMC6276497
- DOI: 10.1155/2018/3839803
Human Cellular Models for the Investigation of Lung Inflammation and Mucus Production in Cystic Fibrosis
Abstract
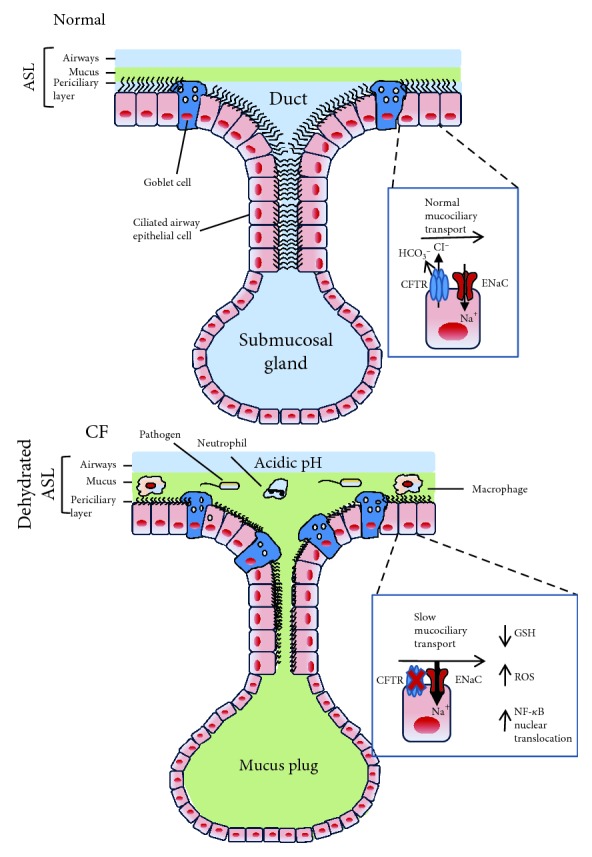
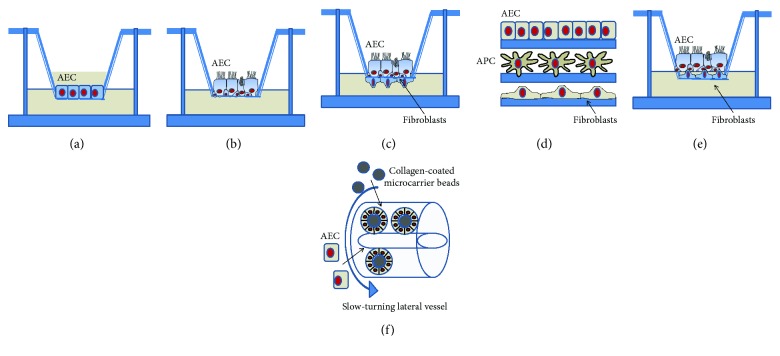
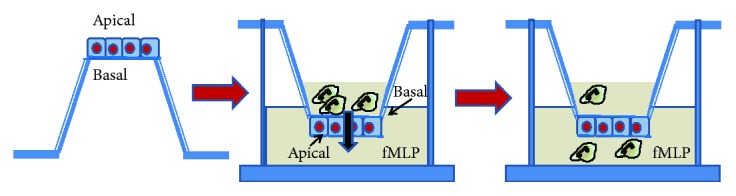
Chronic inflammation, oxidative stress, mucus plugging, airway remodeling, and respiratory infections are the hallmarks of the cystic fibrosis (CF) lung disease. The airway epithelium is central in the innate immune responses to pathogens colonizing the airways, since it is involved in mucociliary clearance, senses the presence of pathogens, elicits an inflammatory response, orchestrates adaptive immunity, and activates mesenchymal cells. In this review, we focus on cellular models of the human CF airway epithelium that have been used for studying mucus production, inflammatory response, and airway remodeling, with particular reference to two- and three-dimensional cultures that better recapitulate the native airway epithelium. Cocultures of airway epithelial cells, macrophages, dendritic cells, and fibroblasts are instrumental in disease modeling, drug discovery, and identification of novel therapeutic targets. Nevertheless, they have to be implemented in the CF field yet. Finally, novel systems hijacking on tissue engineering, including three-dimensional cocultures, decellularized lungs, microfluidic devices, and lung organoids formed in bioreactors, will lead the generation of relevant human preclinical respiratory models a step forward.
Figures
Similar articles
-
Model of mucociliary clearance in cystic fibrosis lungs.J Theor Biol. 2015 May 7;372:81-8. doi: 10.1016/j.jtbi.2015.02.023. Epub 2015 Mar 5. J Theor Biol. 2015. PMID: 25746843
-
CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium.PLoS Biol. 2009 Jul;7(7):e1000155. doi: 10.1371/journal.pbio.1000155. Epub 2009 Jul 21. PLoS Biol. 2009. PMID: 19621064 Free PMC article.
-
Airway mucus, inflammation and remodeling: emerging links in the pathogenesis of chronic lung diseases.Cell Tissue Res. 2017 Mar;367(3):537-550. doi: 10.1007/s00441-016-2562-z. Epub 2017 Jan 20. Cell Tissue Res. 2017. PMID: 28108847 Review.
-
The activin A antagonist follistatin inhibits cystic fibrosis-like lung inflammation and pathology.Immunol Cell Biol. 2015 Jul;93(6):567-74. doi: 10.1038/icb.2015.7. Epub 2015 Mar 10. Immunol Cell Biol. 2015. PMID: 25753271 Free PMC article.
-
Mucus obstruction and inflammation in early cystic fibrosis lung disease: Emerging role of the IL-1 signaling pathway.Pediatr Pulmonol. 2019 Nov;54 Suppl 3:S5-S12. doi: 10.1002/ppul.24462. Pediatr Pulmonol. 2019. PMID: 31715090 Review.
Cited by
-
Harnessing organs-on-a-chip to model tissue regeneration.Cell Stem Cell. 2021 Jun 3;28(6):993-1015. doi: 10.1016/j.stem.2021.05.008. Cell Stem Cell. 2021. PMID: 34087161 Free PMC article. Review.
-
Pathophysiology of Lung Disease and Wound Repair in Cystic Fibrosis.Pathophysiology. 2021 Mar 10;28(1):155-188. doi: 10.3390/pathophysiology28010011. Pathophysiology. 2021. PMID: 35366275 Free PMC article. Review.
-
The Impact of Air Pollution on the Course of Cystic Fibrosis: A Review.Front Physiol. 2022 Jun 2;13:908230. doi: 10.3389/fphys.2022.908230. eCollection 2022. Front Physiol. 2022. PMID: 35721541 Free PMC article. Review.
-
Choice of Differentiation Media Significantly Impacts Cell Lineage and Response to CFTR Modulators in Fully Differentiated Primary Cultures of Cystic Fibrosis Human Airway Epithelial Cells.Cells. 2020 Sep 21;9(9):2137. doi: 10.3390/cells9092137. Cells. 2020. PMID: 32967385 Free PMC article.
-
Human models for COVID-19 research.J Physiol. 2021 Sep;599(18):4255-4267. doi: 10.1113/JP281499. Epub 2021 Aug 17. J Physiol. 2021. PMID: 34287894 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
