

Peak Annotation and Verification Engine for Untargeted LC-MS Metabolomics
- PMID: 30586294
- PMCID: PMC6501219
- DOI: 10.1021/acs.analchem.8b03132
Peak Annotation and Verification Engine for Untargeted LC-MS Metabolomics
Abstract
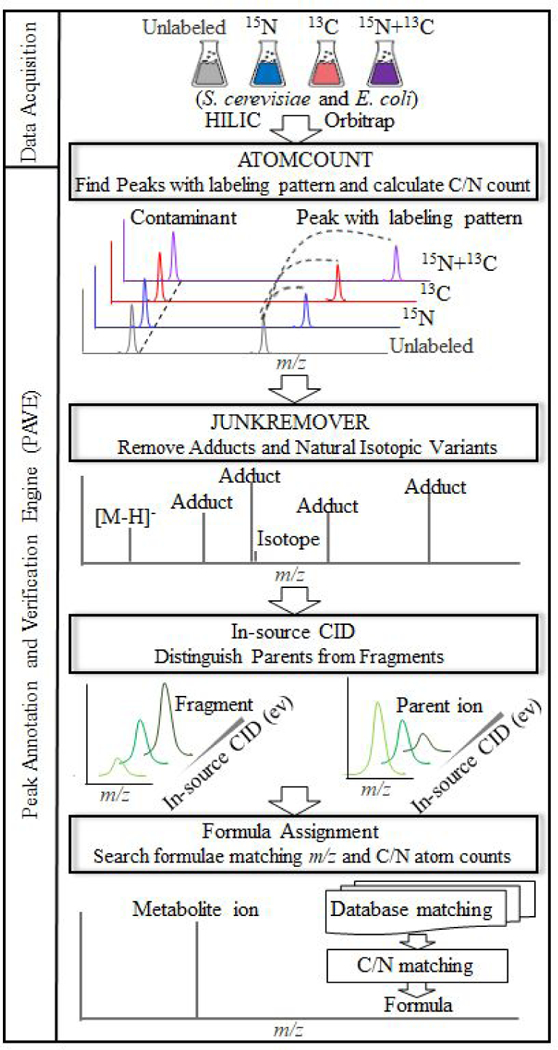
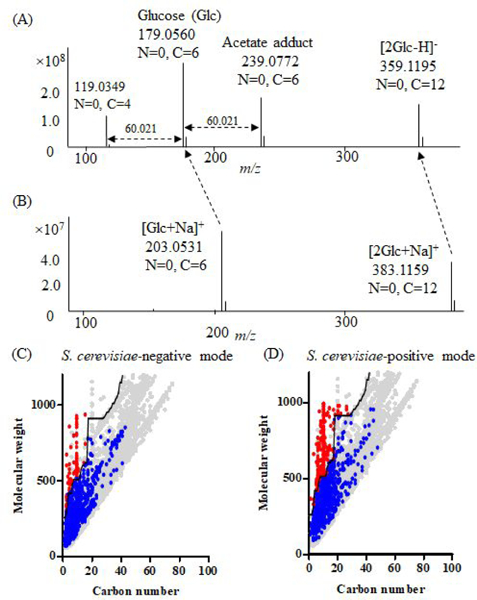
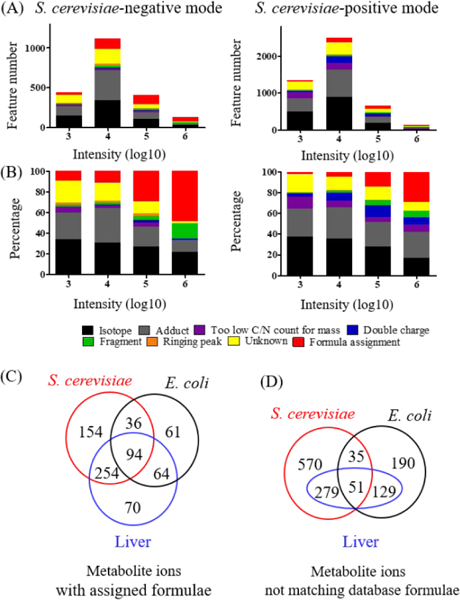
Untargeted metabolomics can detect more than 10 000 peaks in a single LC-MS run. The correspondence between these peaks and metabolites, however, remains unclear. Here, we introduce a Peak Annotation and Verification Engine (PAVE) for annotating untargeted microbial metabolomics data. The workflow involves growing cells in 13C and 15N isotope-labeled media to identify peaks from biological compounds and their carbon and nitrogen atom counts. Improved deisotoping and deadducting are enabled by algorithms that integrate positive mode, negative mode, and labeling data. To distinguish metabolites and their fragments, PAVE experimentally measures the response of each peak to weak in-source collision induced dissociation, which increases the peak intensity for fragments while decreasing it for their parent ions. The molecular formulas of the putative metabolites are then assigned based on database searching using both m/ z and C/N atom counts. Application of this procedure to Saccharomyces cerevisiae and Escherichia coli revealed that more than 80% of peaks do not label, i.e., are environmental contaminants. More than 70% of the biological peaks are isotopic variants, adducts, fragments, or mass spectrometry artifacts yielding ∼2000 apparent metabolites across the two organisms. About 650 match to a known metabolite formula based on m/ z and C/N atom counts, with 220 assigned structures based on MS/MS and/or retention time to match to authenticated standards. Thus, PAVE enables systematic annotation of LC-MS metabolomics data with only ∼4% of peaks annotated as apparent metabolites.
Figures


References
-
- Gowda H; Ivanisevic J; Johnson CH; Kurczy ME; Benton HP; Rinehart D; Nguyen T; Ray J; Kuehl J; Arevalo B; et al. Interactive XCMS Online: Simplifying Advanced Metabolomic Data Processing and Subsequent Statistical Analyses. Analytical Chemistry 2014, 86 (14), 6931–6939. 10.1021/ac500734c. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
