

Epigenetic Silencing of miRNA-338-5p and miRNA-421 Drives SPINK1-Positive Prostate Cancer
- PMID: 30587549
- PMCID: PMC6517268
- DOI: 10.1158/1078-0432.CCR-18-3230
Epigenetic Silencing of miRNA-338-5p and miRNA-421 Drives SPINK1-Positive Prostate Cancer
Abstract
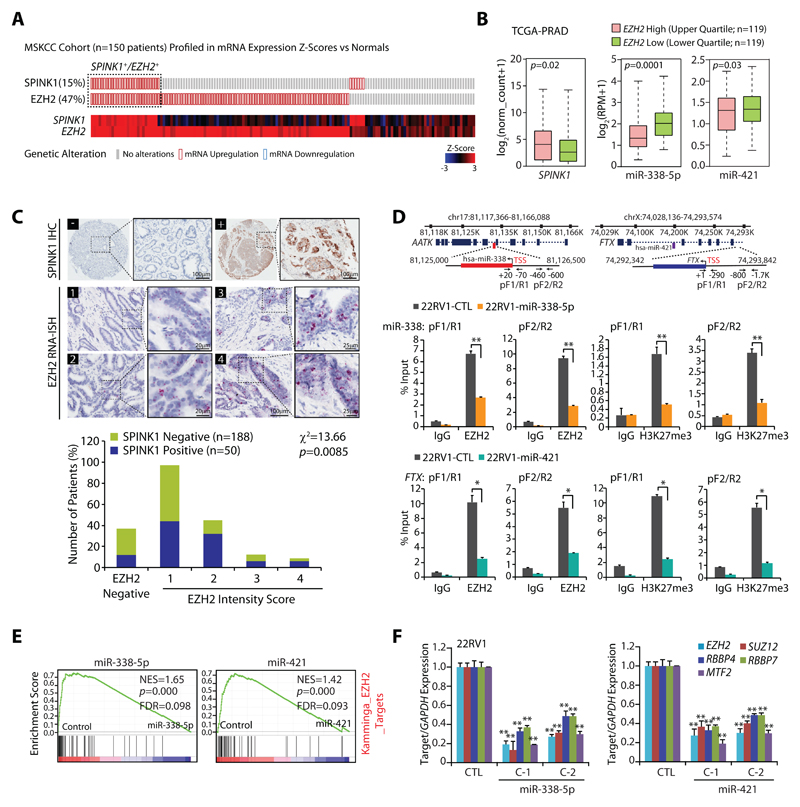
Purpose: Serine peptidase inhibitor, Kazal type-1 (SPINK1) overexpression defines the second most recurrent and aggressive prostate cancer subtype. However, the underlying molecular mechanism and pathobiology of SPINK1 in prostate cancer remains largely unknown.
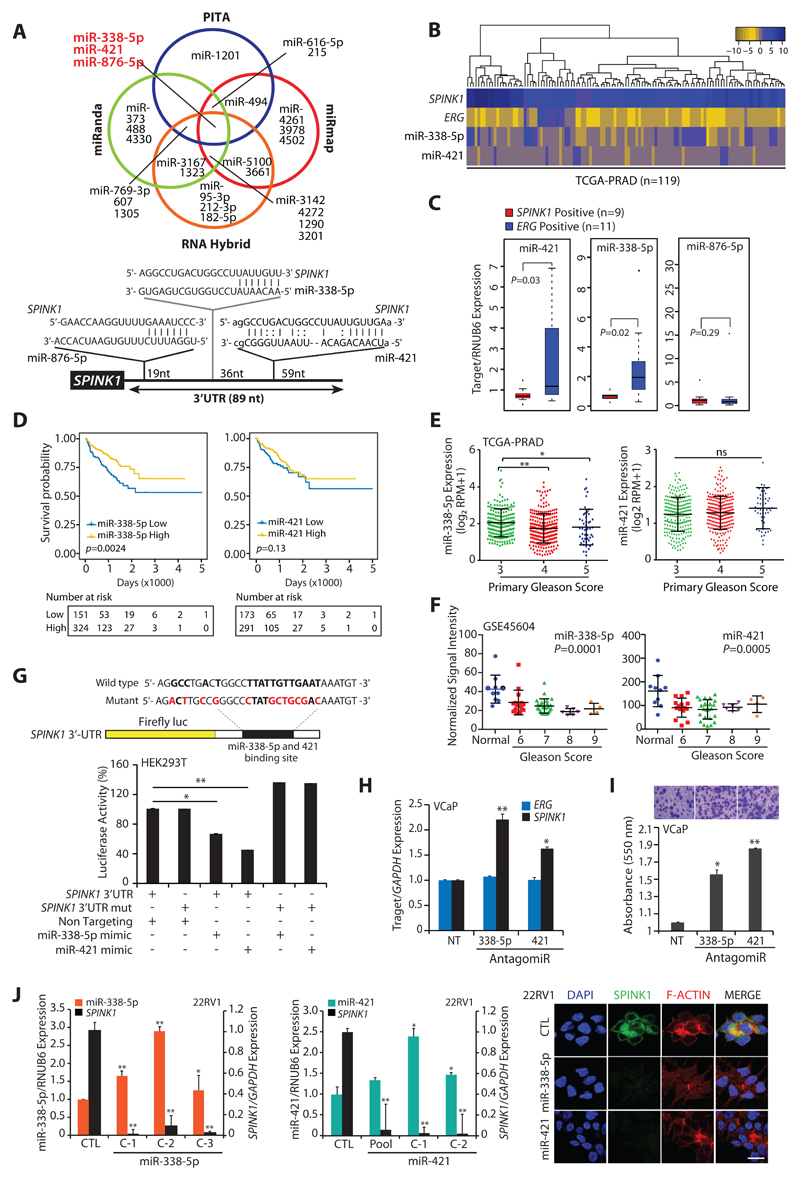
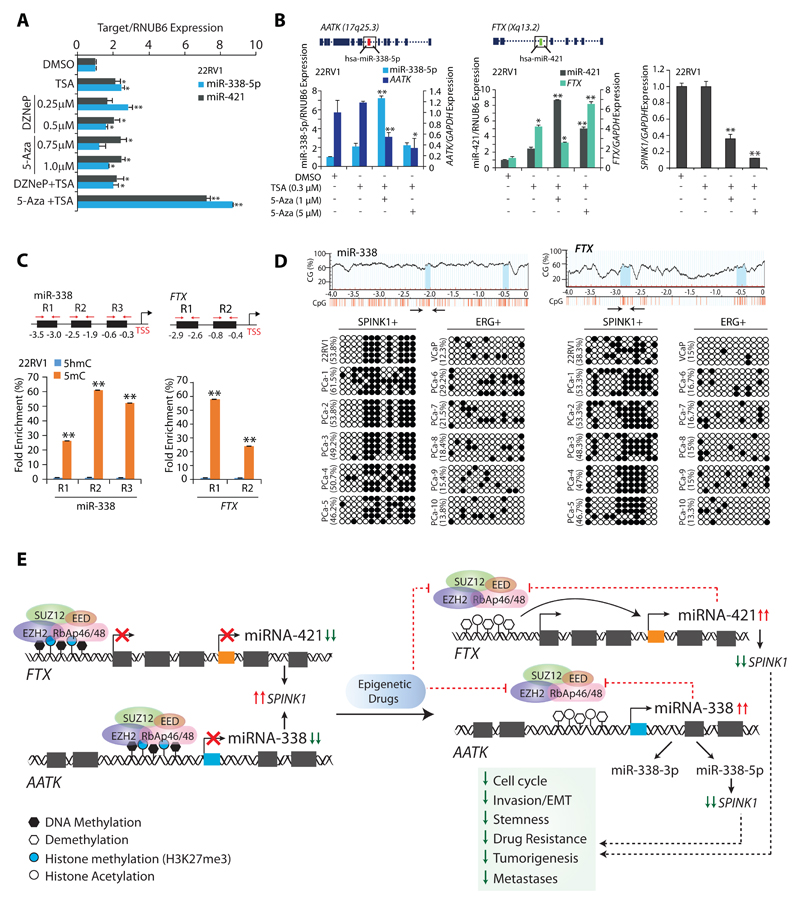
Experimental design: miRNA prediction tools were employed to examine the SPINK1-3'UTR for miRNA binding. Luciferase reporter assays were performed to confirm the SPINK1-3'UTR binding of shortlisted miR-338-5p/miR-421. Furthermore, miR-338-5p/-421-overexpressing cancer cells (SPINK1-positive) were evaluated for oncogenic properties using cell-based functional assays and a mouse xenograft model. Global gene expression profiling was performed to unravel the biological pathways altered by miR-338-5p/-421. IHC and RNA in situ hybridization were carried out on prostate cancer patients' tissue microarray for SPINK1 and EZH2 expression, respectively. Chromatin immunoprecipitation assay was performed to examine EZH2 occupancy on the miR-338-5p/-421-regulatory regions. Bisulfite sequencing and methylated DNA immunoprecipitation were performed on prostate cancer cell lines and patients' specimens.
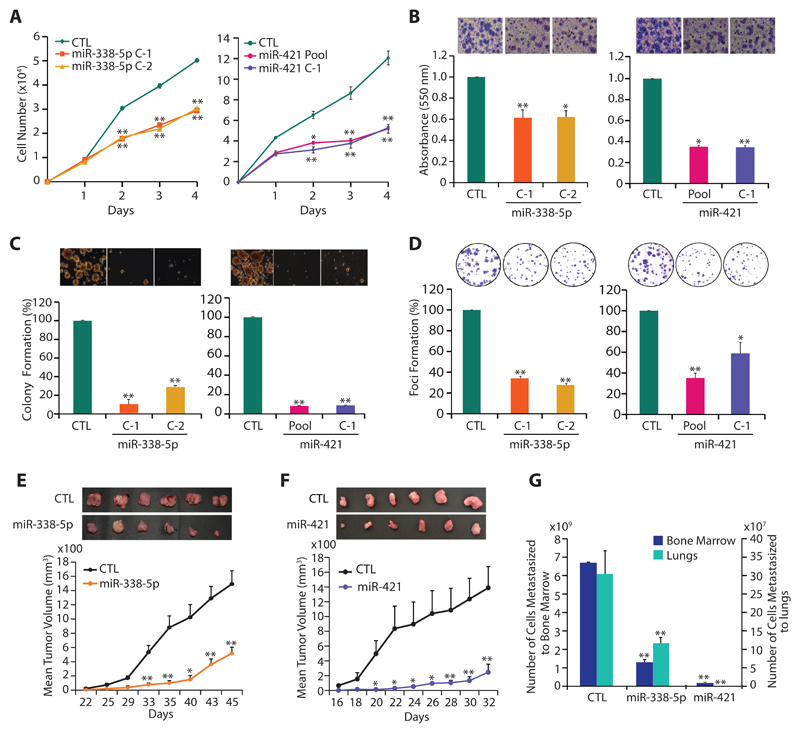
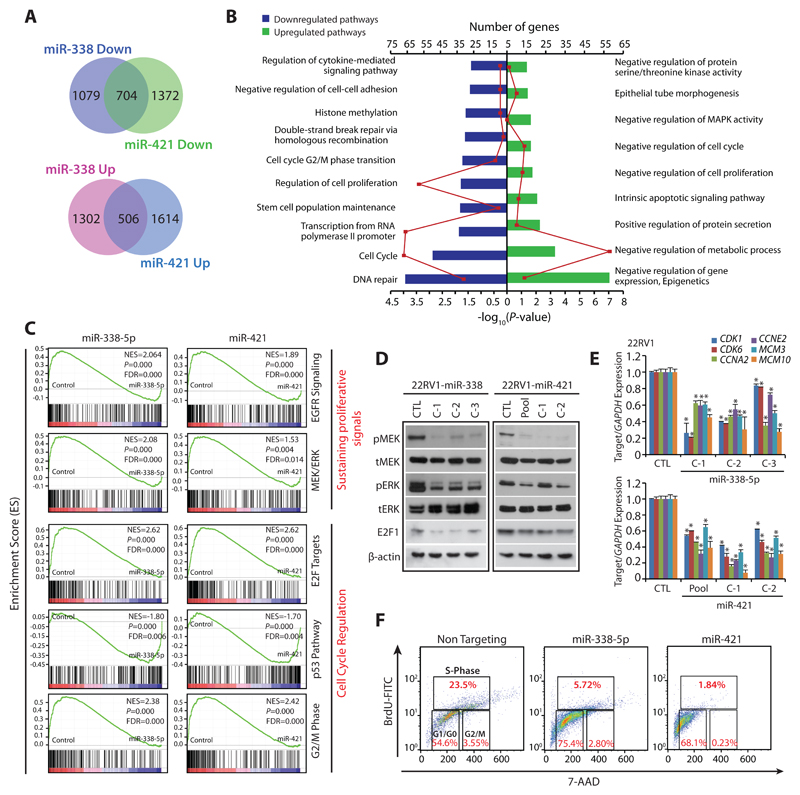
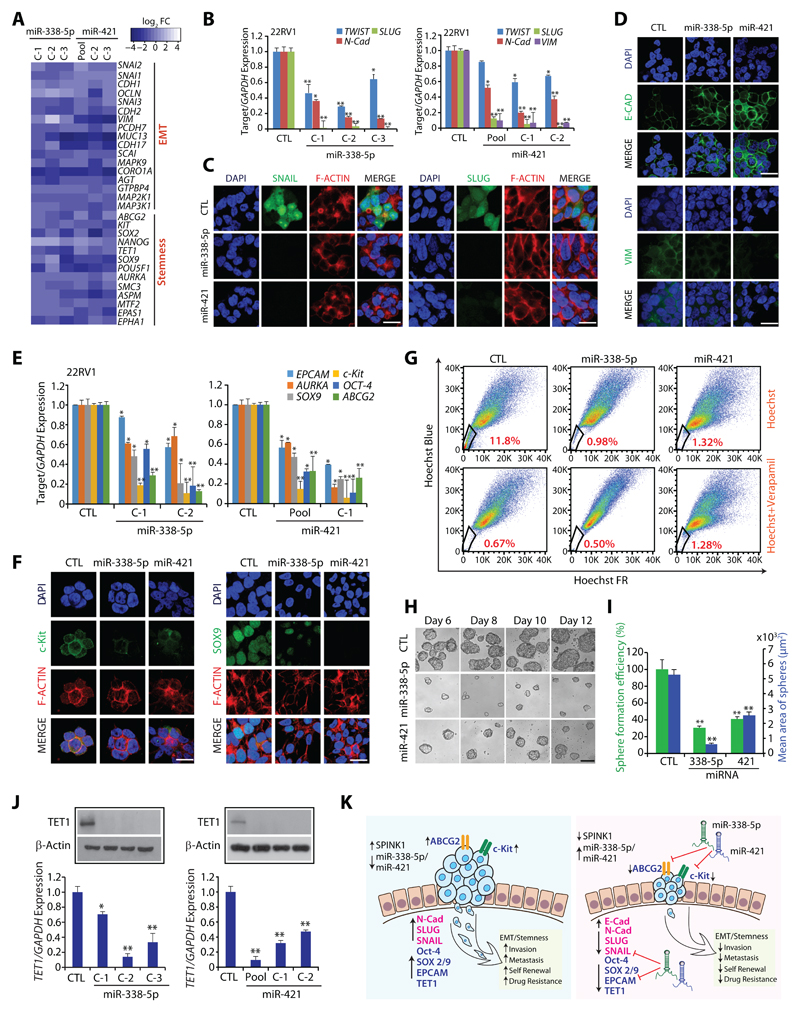
Results: We established a critical role of miRNA-338-5p/-421 in posttranscriptional regulation of SPINK1. Ectopic expression of miRNA-338-5p/-421 in SPINK1-positive cells abrogates oncogenic properties including cell-cycle progression, stemness, and drug resistance, and shows reduced tumor burden and distant metastases in a mouse model. Importantly, we show that patients with SPINK1-positive prostate cancer exhibit increased EZH2 expression, suggesting its role in epigenetic silencing of miRNA-338-5p/-421. Furthermore, presence of CpG dinucleotide DNA methylation marks on the regulatory regions of miR-338-5p/-421 in SPINK1-positive prostate cancer cells and patients' specimens confirms epigenetic silencing.
Conclusions: Our findings revealed that miRNA-338-5p/-421 are epigenetically silenced in SPINK1-positive prostate cancer, although restoring the expression of these miRNAs using epigenetic drugs or synthetic mimics could abrogate SPINK1-mediated oncogenesis.See related commentary by Bjartell, p. 2679.
©2018 American Association for Cancer Research.
Conflict of interest statement
Figures
Comment in
-
New Hope in Prostate Cancer Precision Medicine? miRNA Replacement and Epigenetics.Clin Cancer Res. 2019 May 1;25(9):2679-2681. doi: 10.1158/1078-0432.CCR-19-0061. Epub 2019 Feb 26. Clin Cancer Res. 2019. PMID: 30808772
References
-
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun X-W, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. science. 2005;310(5748):644–8. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
