

Recent advances in the histo-molecular pathology of human prion disease
- PMID: 30588685
- PMCID: PMC8028685
- DOI: 10.1111/bpa.12695
Recent advances in the histo-molecular pathology of human prion disease
Abstract
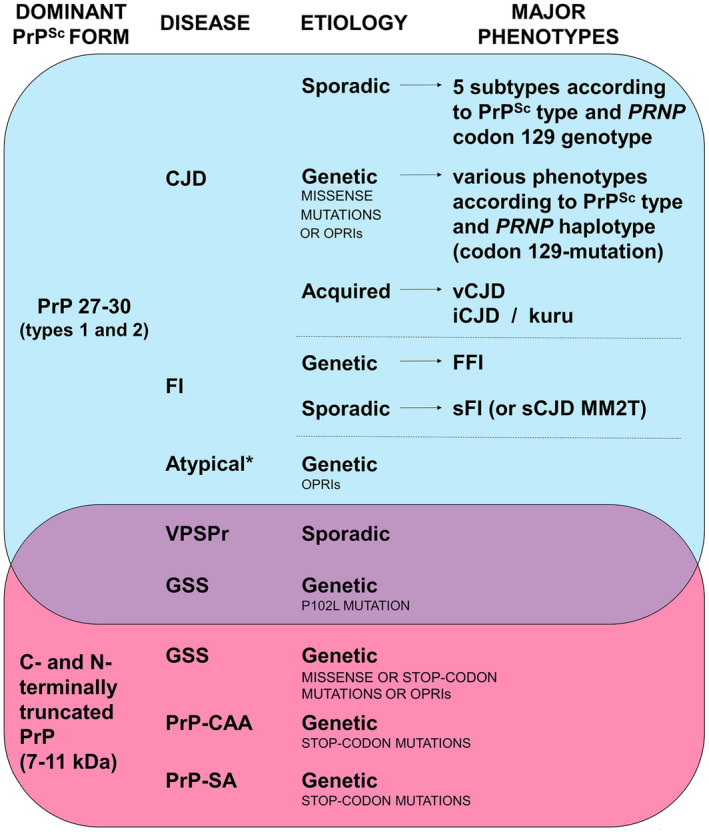
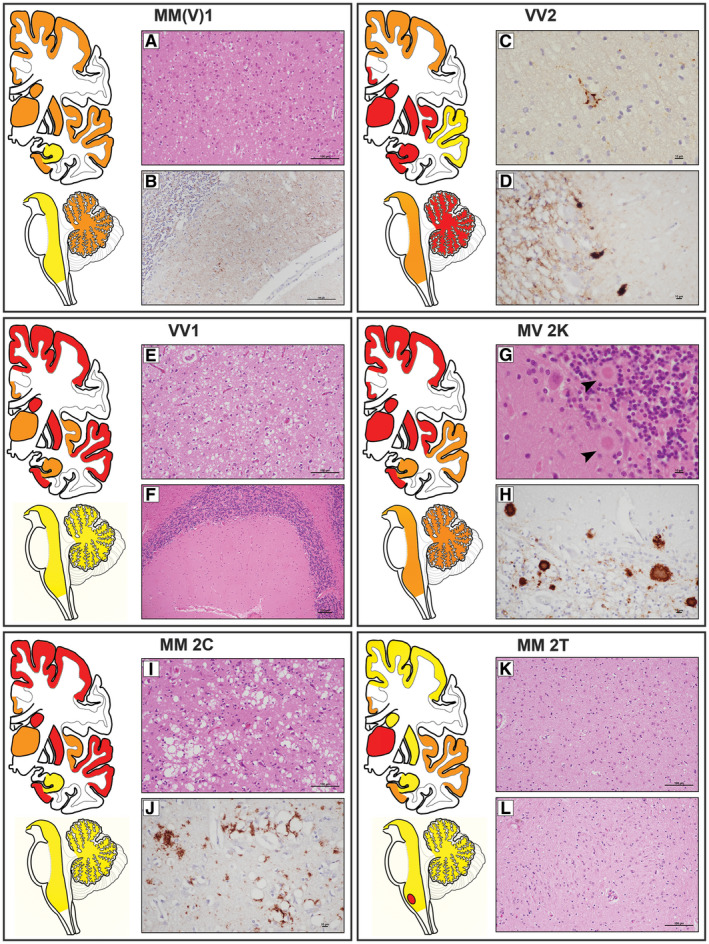
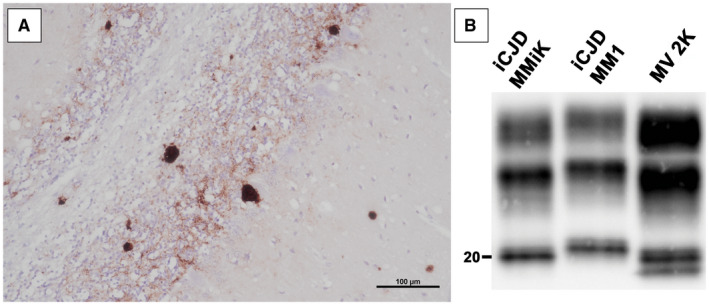
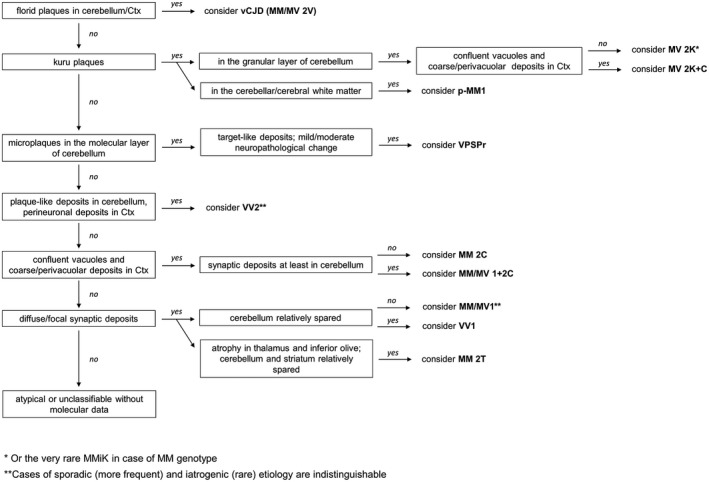
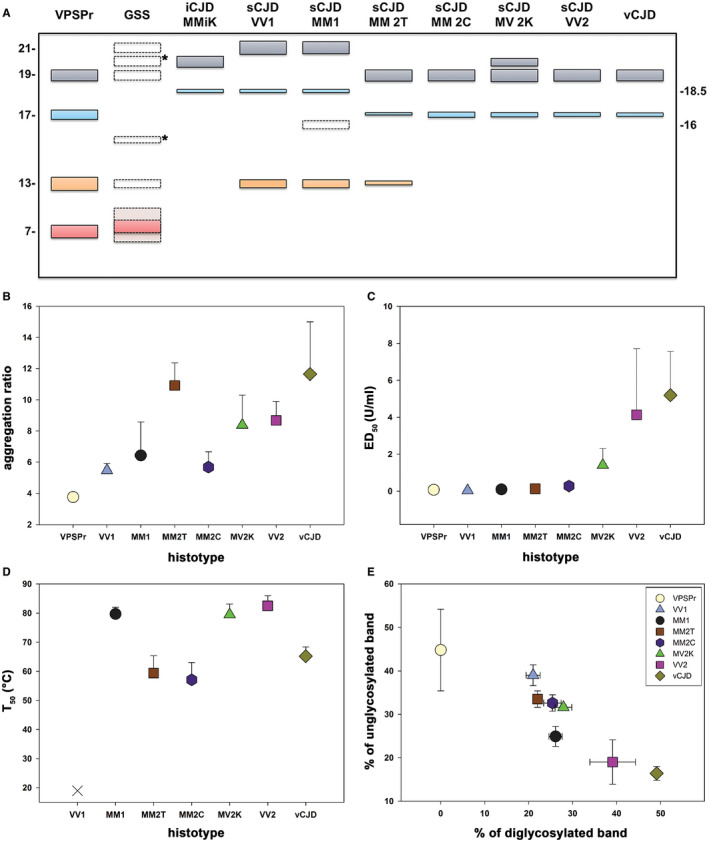
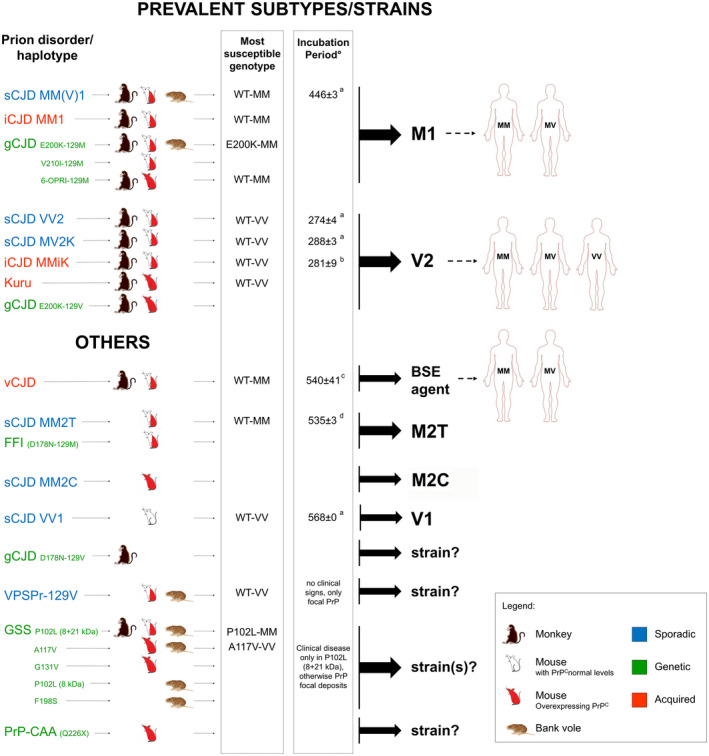
Prion diseases are progressive neurodegenerative disorders affecting humans and other mammalian species. The term prion, originally put forward to propose the concept that a protein could be infectious, refers to PrPSc , a misfolded isoform of the cellular prion protein (PrPC ) that represents the pathogenetic hallmark of these disorders. The discovery that other proteins characterized by misfolding and seeded aggregation can spread from cell to cell, similarly to PrPSc , has increased interest in prion diseases. Among neurodegenerative disorders, however, prion diseases distinguish themselves for the broader phenotypic spectrum, the fastest disease progression and the existence of infectious forms that can be transmitted through the exposure to diseased tissues via ingestion, injection or transplantation. The main clinicopathological phenotypes of human prion disease include Creutzfeldt-Jakob disease, by far the most common, fatal insomnia, variably protease-sensitive prionopathy, and Gerstmann-Sträussler-Scheinker disease. However, clinicopathological manifestations extend even beyond those predicted by this classification. Because of their transmissibility, the phenotypic diversity of prion diseases can also be propagated into syngenic hosts as prion strains with distinct characteristics, such as incubation period, pattern of PrPSc distribution and regional severity of histopathological changes in the brain. Increasing evidence indicates that different PrPSc conformers, forming distinct ordered aggregates, encipher the phenotypic variants related to prion strains. In this review, we summarize the most recent advances concerning the histo-molecular pathology of human prion disease focusing on the phenotypic spectrum of the disease including co-pathologies, the characterization of prion strains by experimental transmission and their correlation with the physicochemical properties of PrPSc aggregates.
Keywords: Creutzfeldt-Jakob disease; Gerstmann-Sträussler-Scheinker disease; amyloidosis; fatal insomnia; human prions; neurodegenerative dementia; prion strains.
© 2019 International Society of Neuropathology.
Figures
References
-
- Abu‐Rumeileh S, Redaelli V, Baiardi S, Mackenzie G, Windl O, Ritchie DL et al (2018) Sporadic fatal insomnia in Europe: phenotypic features and diagnostic challenges. Ann Neurol 84:347–360. - PubMed
-
- Aguzzi A, Heikenwalder M, Polymenidou M (2007) Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 8:552–561. - PubMed
-
- Almer G, Hainfellner JA, Brücke T, Jellinger K, Kleinert R, Bayer G et al (1999) Fatal familial insomnia: a new Austrian family. Brain 122(Pt 1):5–16. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
