

PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer
- PMID: 30589644
- PMCID: PMC6391116
- DOI: 10.1172/JCI123319
PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer
Abstract
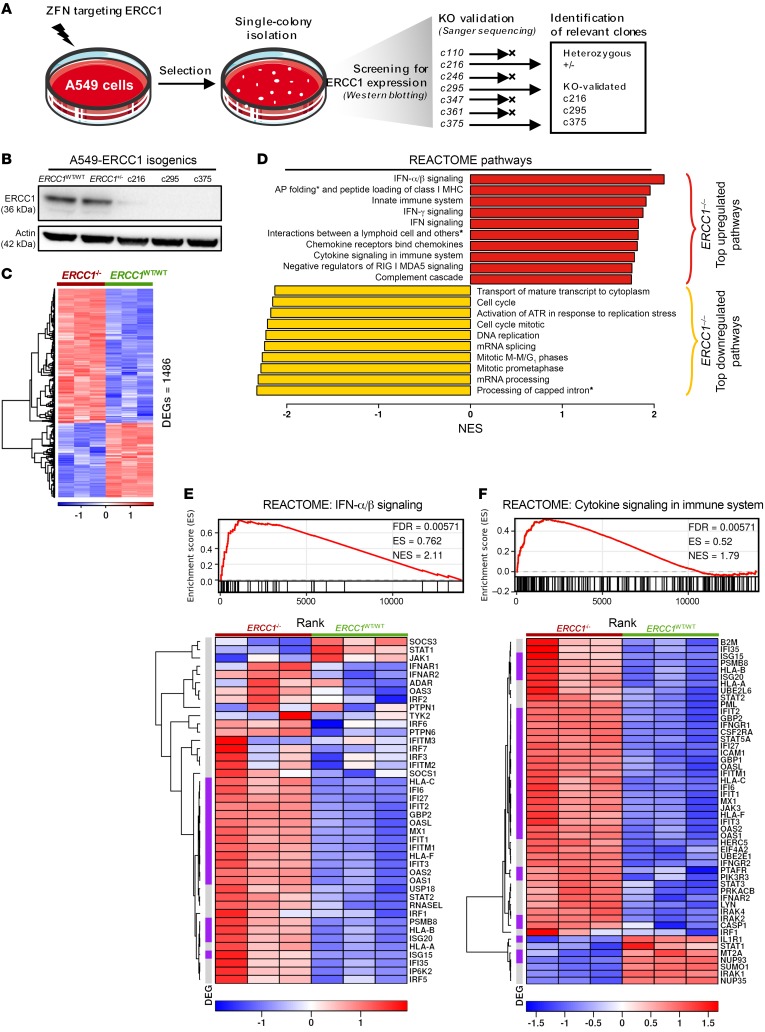
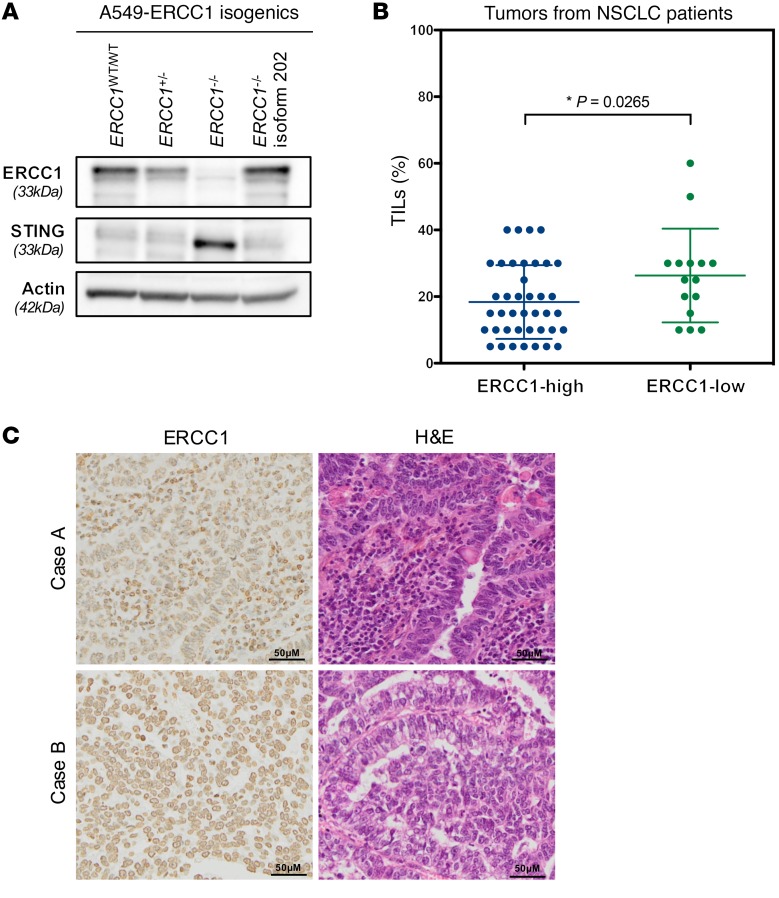
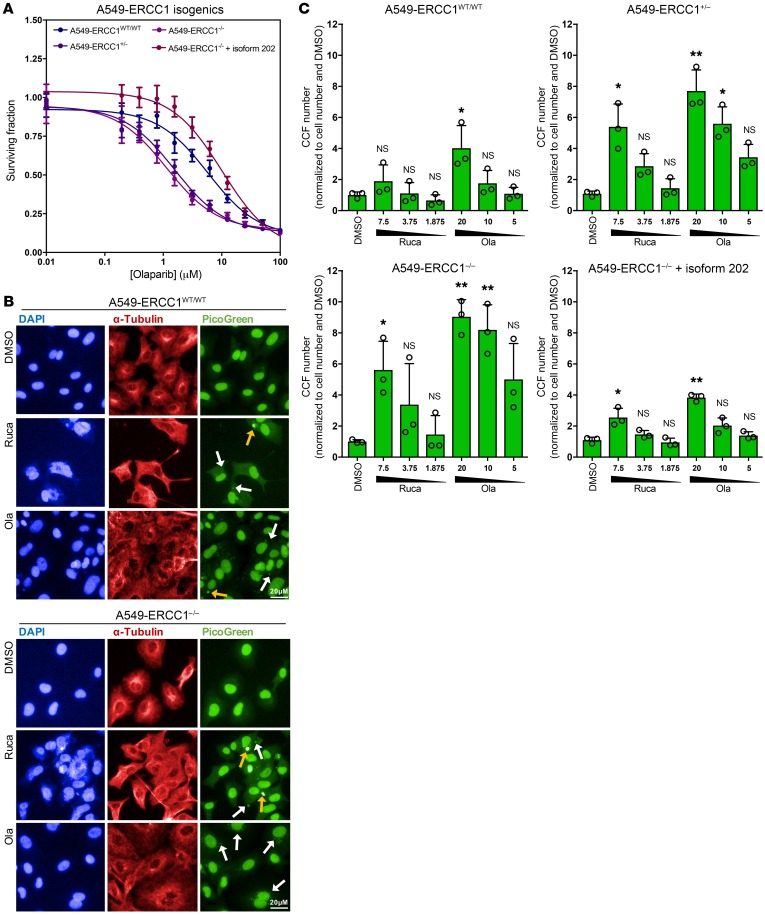
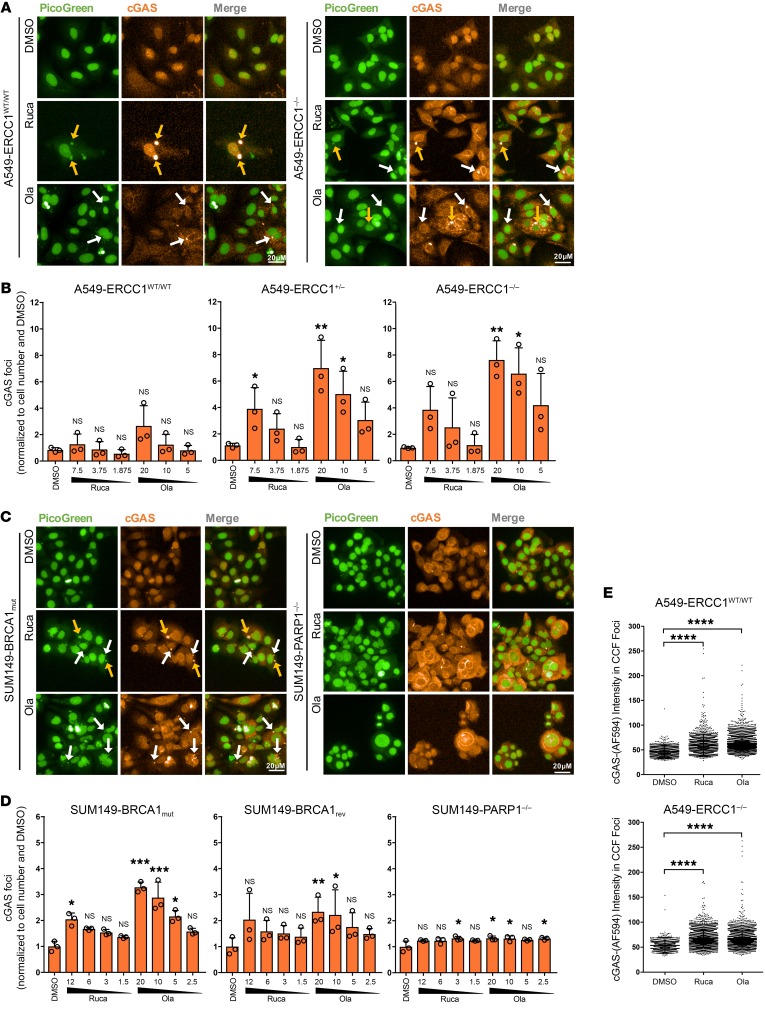
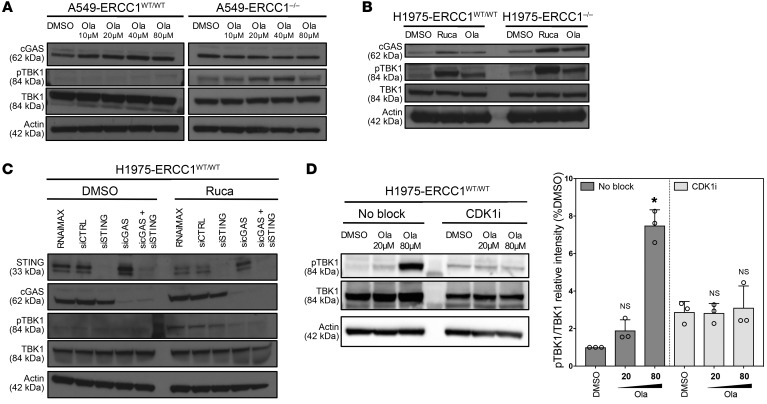
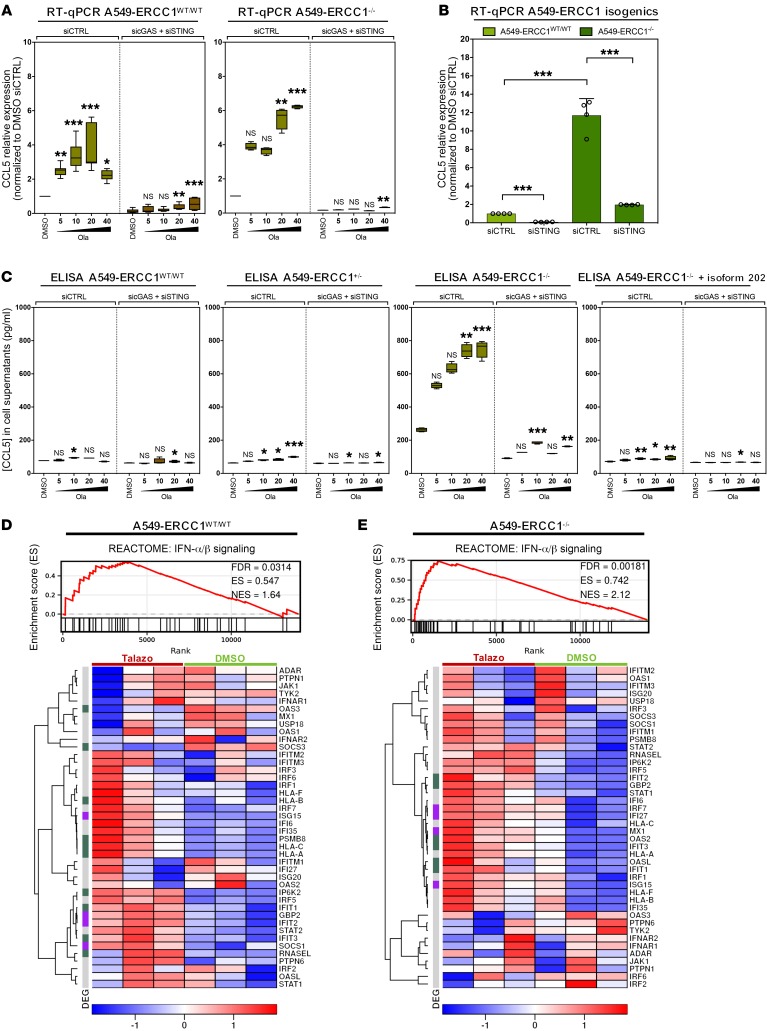
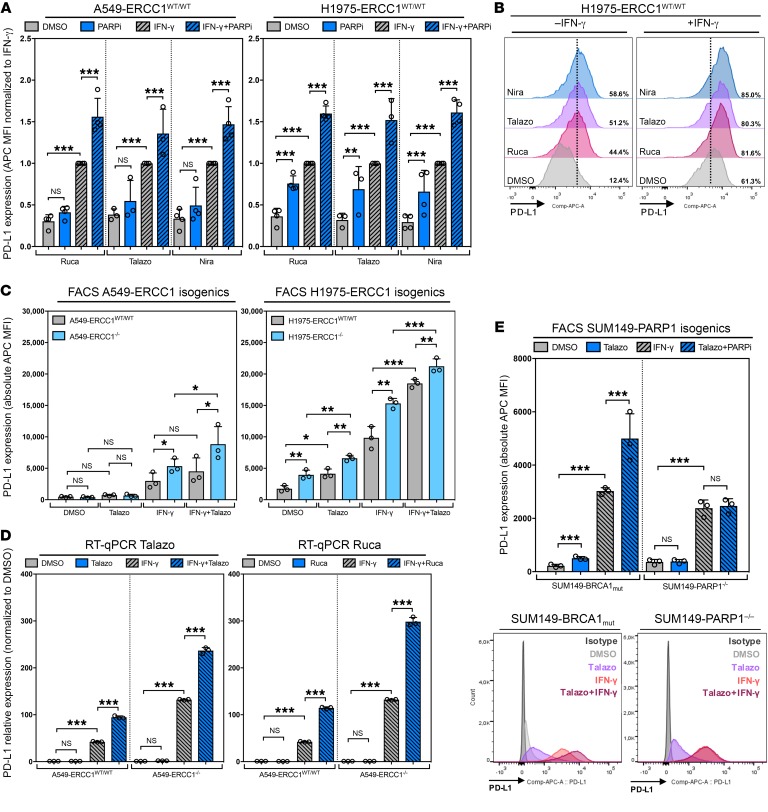
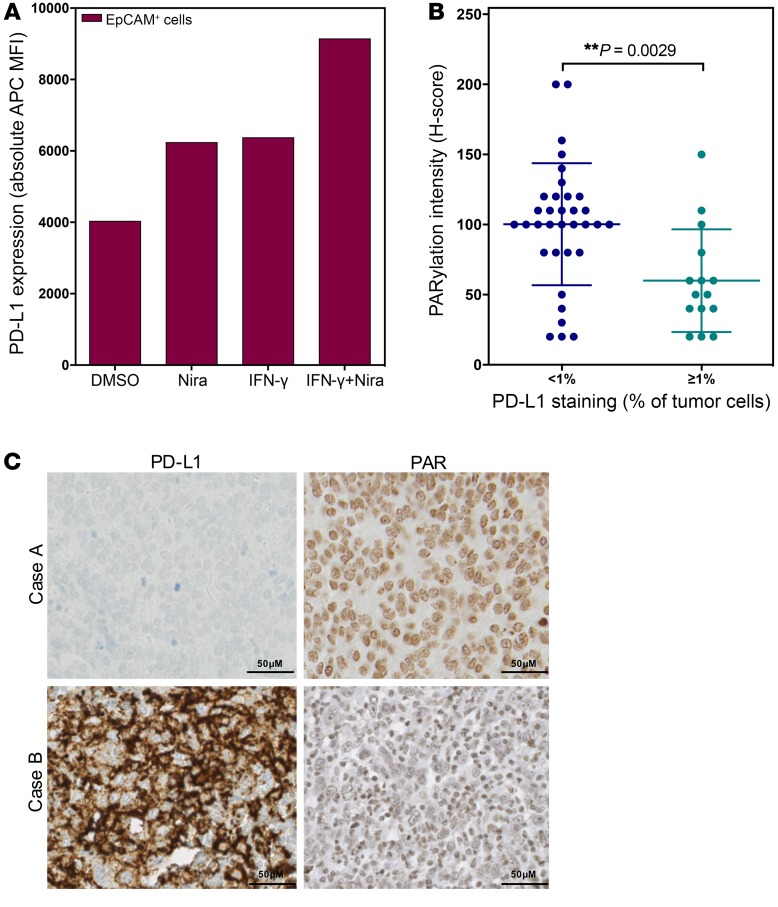
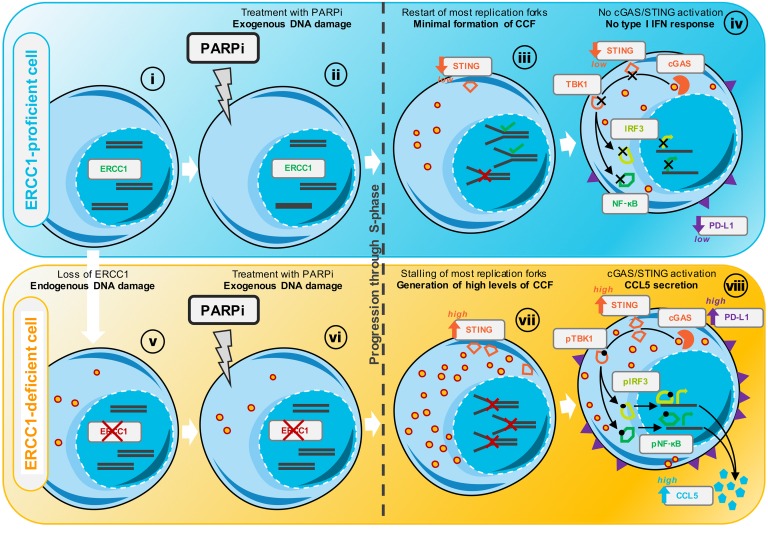
The cyclic GMP-AMP synthase/stimulator of IFN genes (cGAS/STING) pathway detects cytosolic DNA to activate innate immune responses. Poly(ADP-ribose) polymerase inhibitors (PARPi) selectively target cancer cells with DNA repair deficiencies such as those caused by BRCA1 mutations or ERCC1 defects. Using isogenic cell lines and patient-derived samples, we showed that ERCC1-defective non-small cell lung cancer (NSCLC) cells exhibit an enhanced type I IFN transcriptomic signature and that low ERCC1 expression correlates with increased lymphocytic infiltration. We demonstrated that clinical PARPi, including olaparib and rucaparib, have cell-autonomous immunomodulatory properties in ERCC1-defective NSCLC and BRCA1-defective triple-negative breast cancer (TNBC) cells. Mechanistically, PARPi generated cytoplasmic chromatin fragments with characteristics of micronuclei; these were found to activate cGAS/STING, downstream type I IFN signaling, and CCL5 secretion. Importantly, these effects were suppressed in PARP1-null TNBC cells, suggesting that this phenotype resulted from an on-target effect of PARPi on PARP1. PARPi also potentiated IFN-γ-induced PD-L1 expression in NSCLC cell lines and in fresh patient tumor cells; this effect was enhanced in ERCC1-deficient contexts. Our data provide a preclinical rationale for using PARPi as immunomodulatory agents in appropriately molecularly selected populations.
Keywords: Cellular immune response; DNA repair; Lung cancer; Oncology.
Conflict of interest statement
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
