

Population structure in genetic studies: Confounding factors and mixed models
- PMID: 30589851
- PMCID: PMC6307707
- DOI: 10.1371/journal.pgen.1007309
Population structure in genetic studies: Confounding factors and mixed models
Abstract
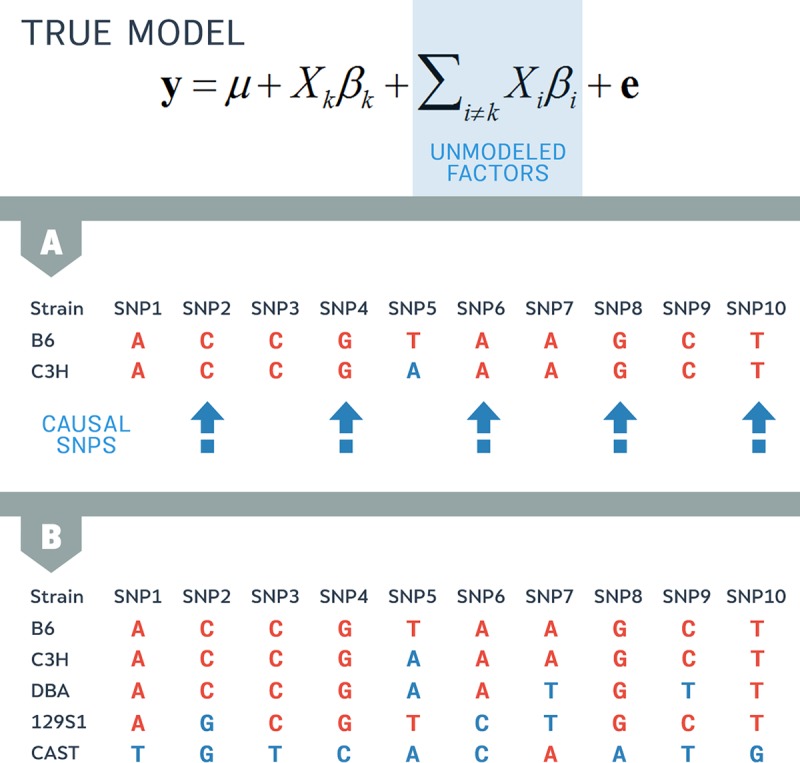
A genome-wide association study (GWAS) seeks to identify genetic variants that contribute to the development and progression of a specific disease. Over the past 10 years, new approaches using mixed models have emerged to mitigate the deleterious effects of population structure and relatedness in association studies. However, developing GWAS techniques to accurately test for association while correcting for population structure is a computational and statistical challenge. Using laboratory mouse strains as an example, our review characterizes the problem of population structure in association studies and describes how it can cause false positive associations. We then motivate mixed models in the context of unmodeled factors.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures










References
-
- Stram DO. Design, analysis, and interpretation of genome-wide association scans. New York: Springer; 2014. xv, 334 pages p.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
