

Discerning novel splice junctions derived from RNA-seq alignment: a deep learning approach
- PMID: 30591034
- PMCID: PMC6307148
- DOI: 10.1186/s12864-018-5350-1
Discerning novel splice junctions derived from RNA-seq alignment: a deep learning approach
Abstract
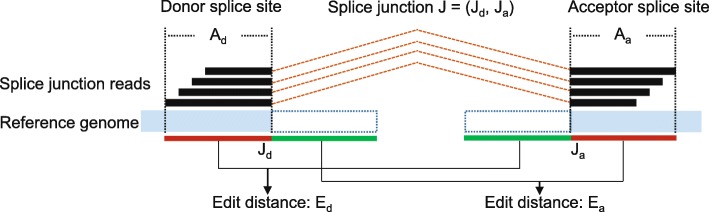
Background: Exon splicing is a regulated cellular process in the transcription of protein-coding genes. Technological advancements and cost reductions in RNA sequencing have made quantitative and qualitative assessments of the transcriptome both possible and widely available. RNA-seq provides unprecedented resolution to identify gene structures and resolve the diversity of splicing variants. However, currently available ab initio aligners are vulnerable to spurious alignments due to random sequence matches and sample-reference genome discordance. As a consequence, a significant set of false positive exon junction predictions would be introduced, which will further confuse downstream analyses of splice variant discovery and abundance estimation.
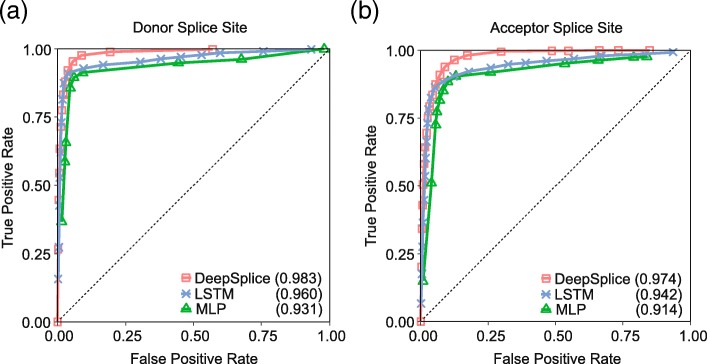
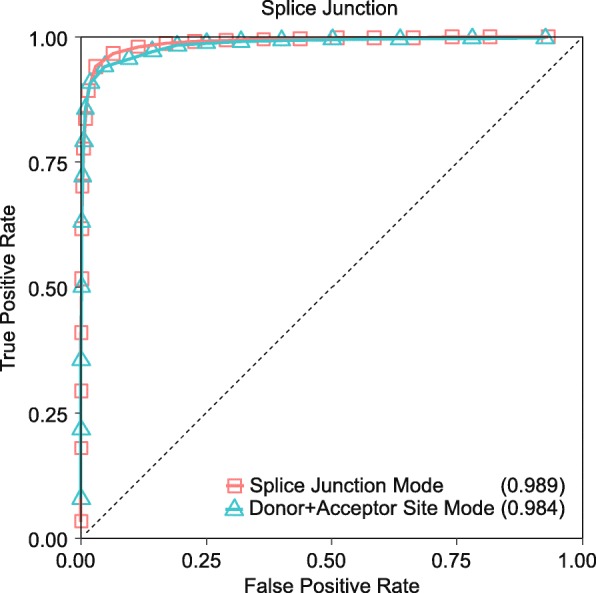
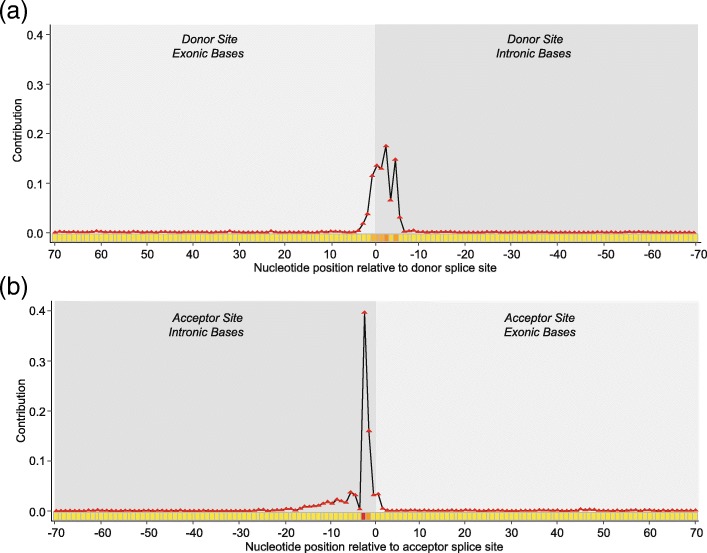
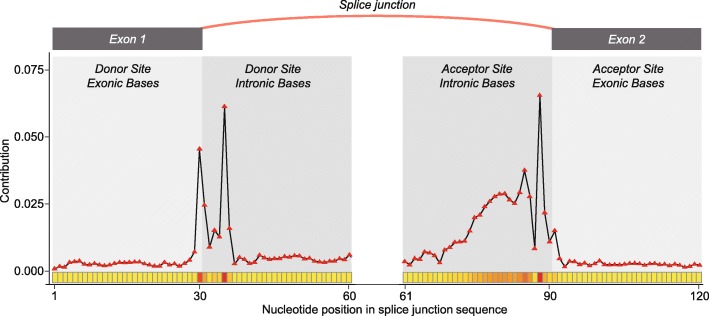
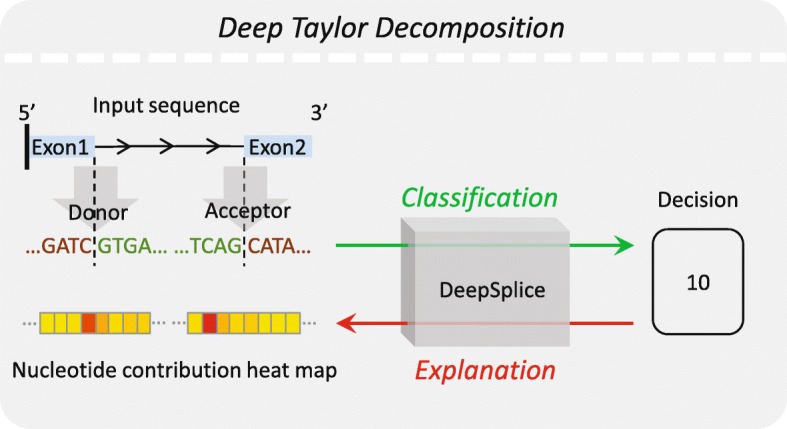
Results: In this work, we present a deep learning based splice junction sequence classifier, named DeepSplice, which employs convolutional neural networks to classify candidate splice junctions. We show (I) DeepSplice outperforms state-of-the-art methods for splice site classification when applied to the popular benchmark dataset HS3D, (II) DeepSplice shows high accuracy for splice junction classification with GENCODE annotation, and (III) the application of DeepSplice to classify putative splice junctions generated by Rail-RNA alignment of 21,504 human RNA-seq data significantly reduces 43 million candidates into around 3 million highly confident novel splice junctions.
Conclusions: A model inferred from the sequences of annotated exon junctions that can then classify splice junctions derived from primary RNA-seq data has been implemented. The performance of the model was evaluated and compared through comprehensive benchmarking and testing, indicating a reliable performance and gross usability for classifying novel splice junctions derived from RNA-seq alignment.
Keywords: Deep learning; Exon splicing; RNA-seq; Splice junction.
Conflict of interest statement
Ethics approval and consent to participate
No permission was required from the ethics committee as the project did not involve testing of human, animal or endangered plant species subjects.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
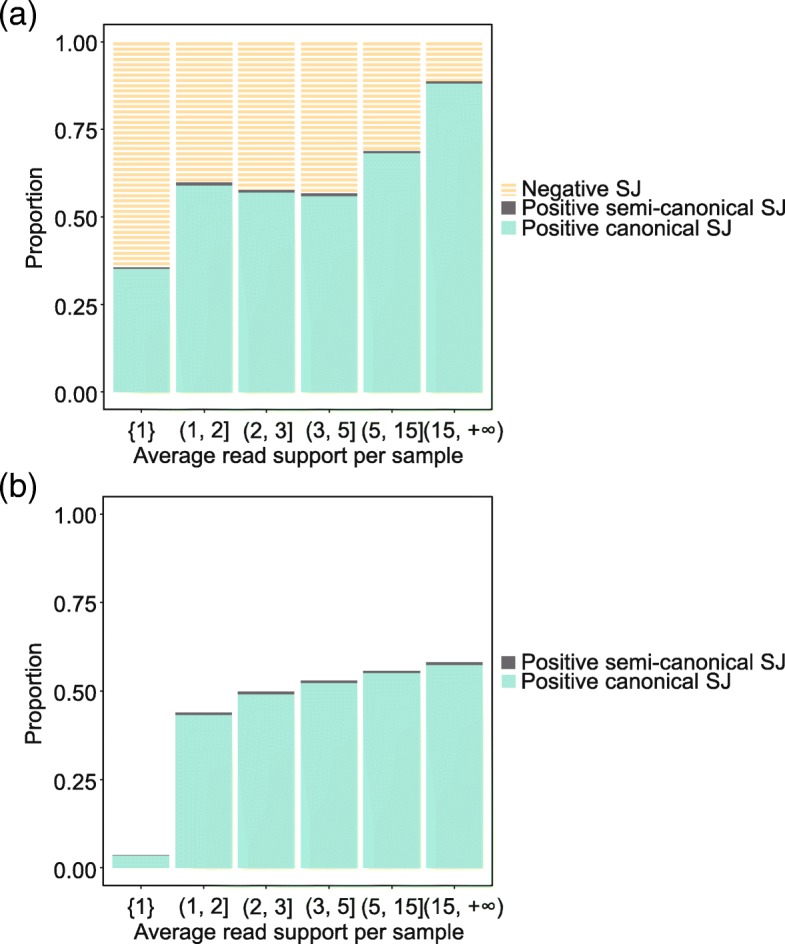
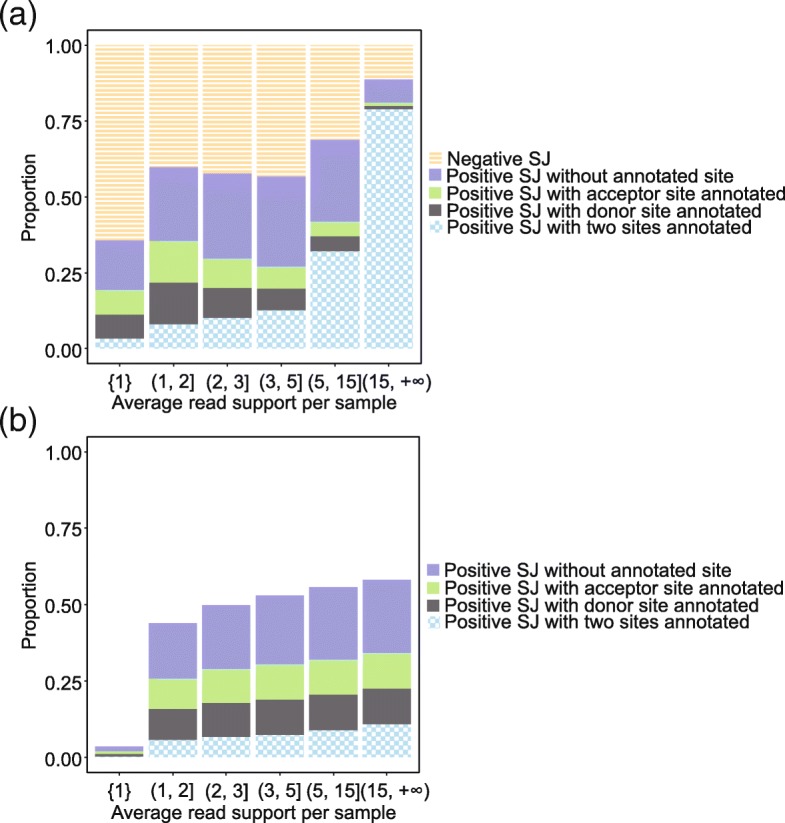
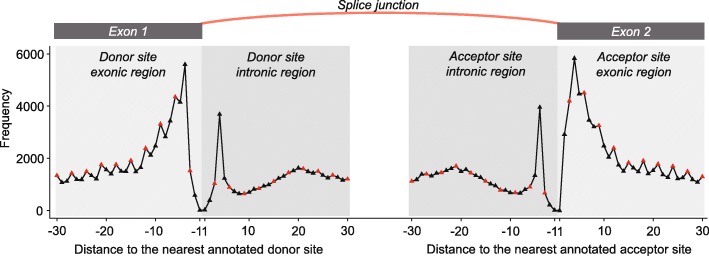
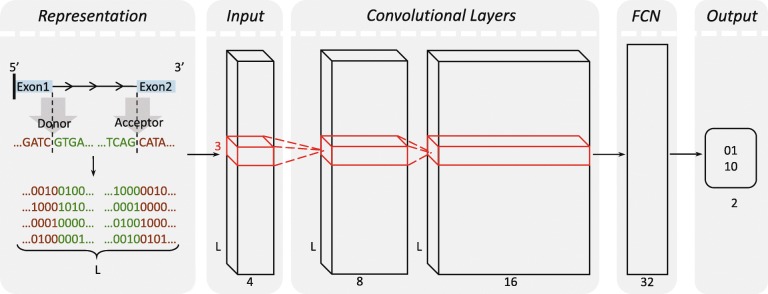
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
