

Homozygous TBC1 domain-containing kinase (TBCK) mutation causes a novel lysosomal storage disease - a new type of neuronal ceroid lipofuscinosis (CLN15)?
- PMID: 30591081
- PMCID: PMC6307319
- DOI: 10.1186/s40478-018-0646-6
Homozygous TBC1 domain-containing kinase (TBCK) mutation causes a novel lysosomal storage disease - a new type of neuronal ceroid lipofuscinosis (CLN15)?
Abstract
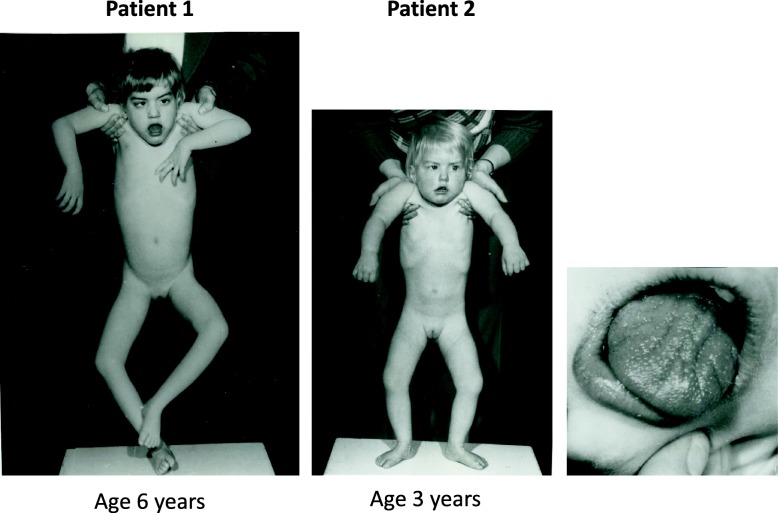
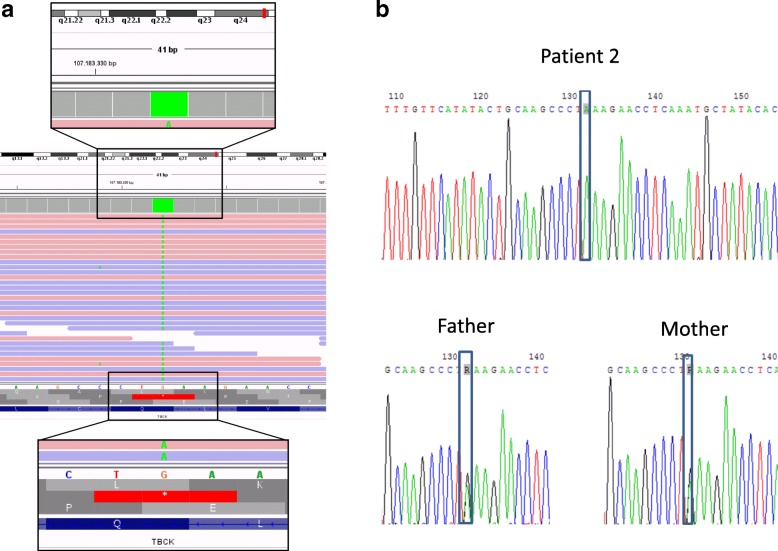
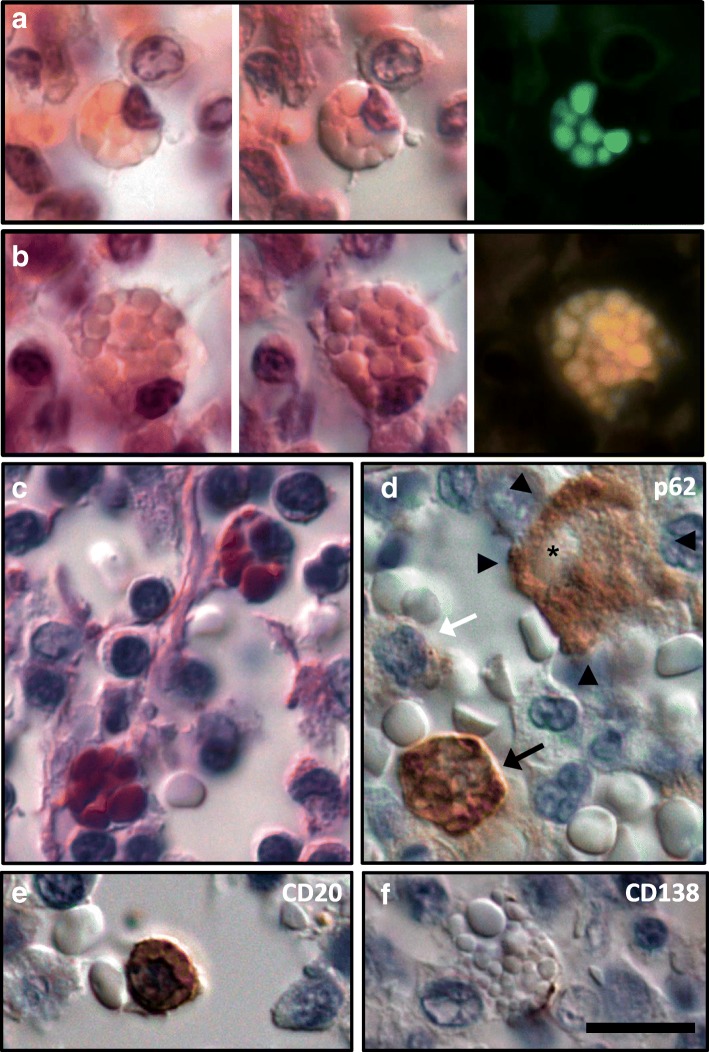
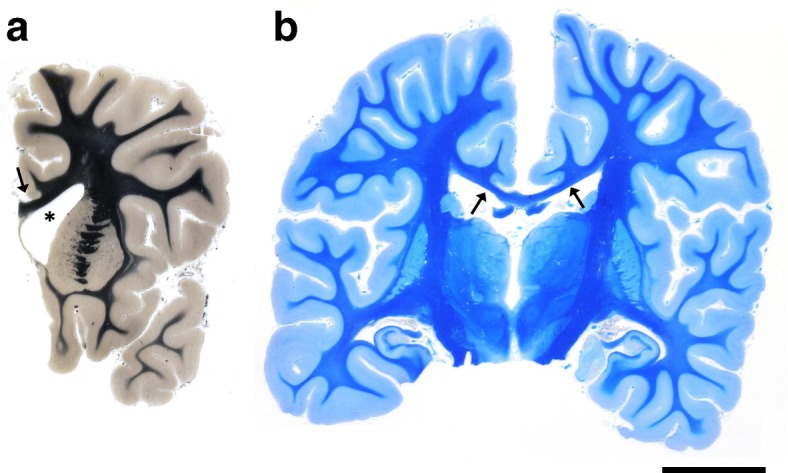
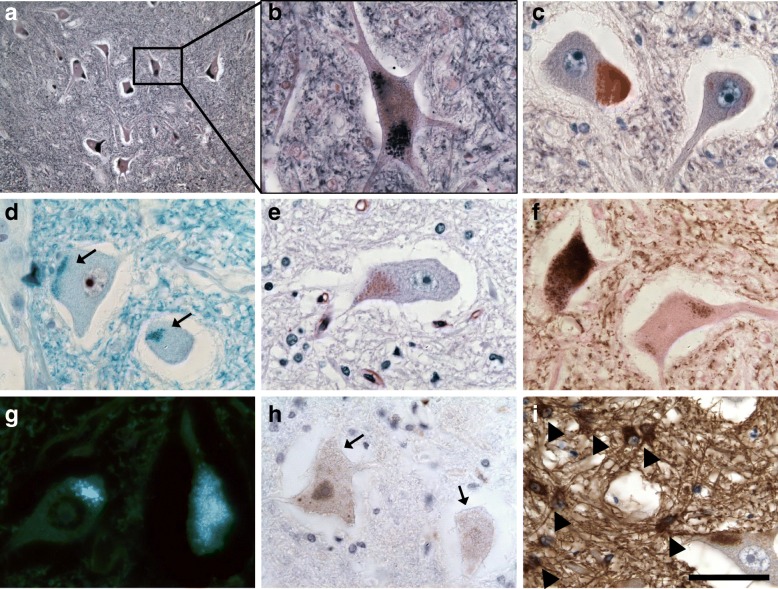
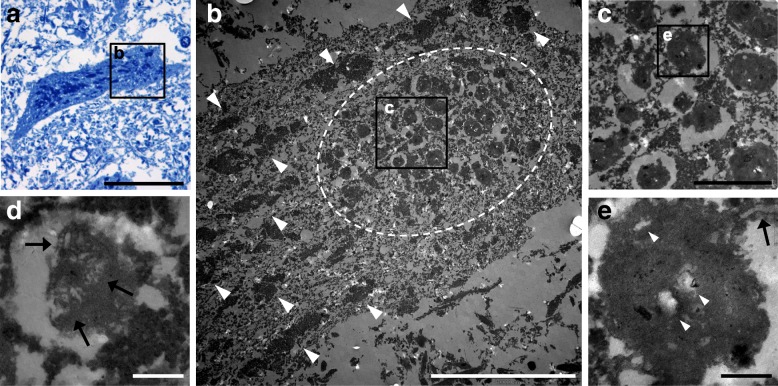
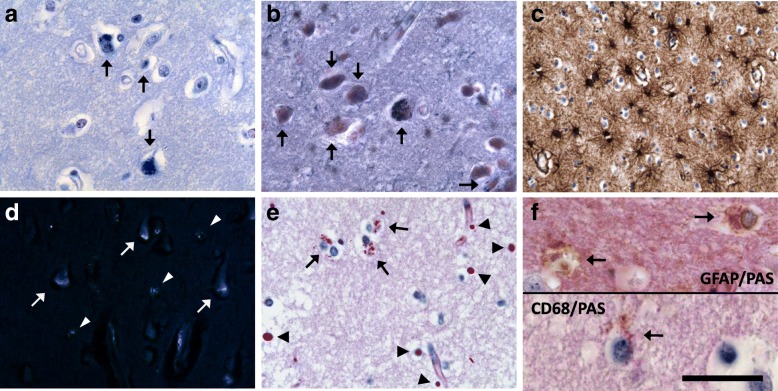
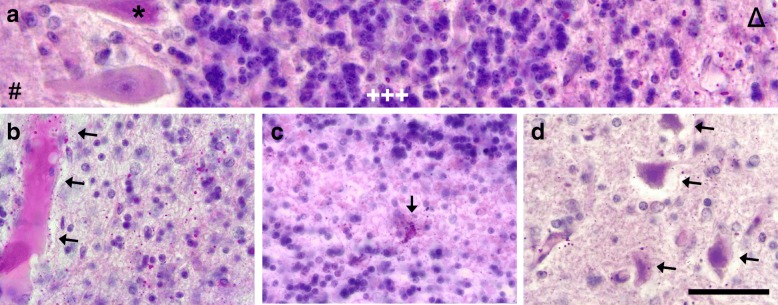
Homozygous mutation of TBC1 domain-containing kinase (TBCK) is the cause of a very recently defined severe childhood disorder, which is characterized by severe hypotonia, global developmental delay, intellectual disability, epilepsy, characteristic facies and premature death. The link between TBCK loss of function and symptoms in patients with TBCK deficiency disorder (TBCK-DD) remains elusive. Here we demonstrate for the first time the histopathological characteristics of TBCK deficiency consisting of 1) a widespread and massive accumulation of lipofuscin storage material in neurons of the central nervous system without notable neuronal degeneration, 2) storage deposits in few astrocytes, 3) carbohydrate-rich deposits in brain, spleen and liver and 4) vacuolated lymphocytes. Biochemical examinations ruled out more than 20 known lysosomal storage diseases. These investigations strikingly uncover TBCK-DD as a novel type of lysosomal storage disease which is characterized by different storage products rather than one specific type of accumulated material. Due to the clear predominance of intraneuronal lipofuscin storage material and the characteristic clinical presentation we propose to classify this disease as a new subtype of neuronal ceroid lipofuscinosis (CLN15). Our results and previous reports suggest an autophagosomal-lysosomal dysfunction caused by enhanced mTORC1-mediated autophagosome formation and reduced Rab-mediated autophagosome-lysosome fusion, thus disclosing potential novel targets for therapeutic approaches in TBCK-DD.
Keywords: Autophagy; Central nervous system (CNS); Infantile muscular hypotonia with psychomotor retardation and characteristic facies 3 (IHPRF3); Mammalian target of rapamycin (mTOR); Rab; Vacuolated lymphocytes.
Conflict of interest statement
Ethics approval and consent to participate
All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Consent for publication
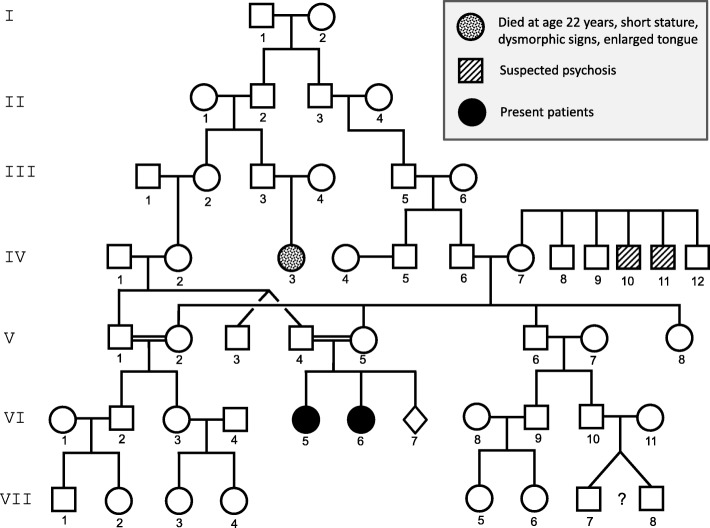
Informed consent was obtained from the parents of the siblings for whom identifying information is included in this article.
Competing interests
The authors declare that they have no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Alazami AM, Patel N, Shamseldin HE, Anazi S, Al-Dosari MS, Alzahrani F, Hijazi H, Alshammari M, Aldahmesh MA, Salih MA, Faqeih E, Alhashem A, Bashiri FA, Al-Owain M, Kentab AY, Sogaty S, Al Tala S, Temsah MH, Tulbah M, Aljelaify RF, Alshahwan SA, Seidahmed MZ, Alhadid AA, Aldhalaan H, AlQallaf F, Kurdi W, Alfadhel M, Babay Z, Alsogheer M, Kaya N, Al-Hassnan ZN, Abdel-Salam GM, Al-Sannaa N, Al Mutairi F, El Khashab HY, Bohlega S, Jia X, Nguyen HC, Hammami R, Adly N, Mohamed JY, Abdulwahab F, Ibrahim N, Naim EA, Al-Younes B, Meyer BF, Hashem M, Shaheen R, Xiong Y, Abouelhoda M, Aldeeri AA, Monies DM, Alkuraya FS. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015;10:148–161. doi: 10.1016/j.celrep.2014.12.015. - DOI - PubMed
-
- Bhoj EJ, Li D, Harr M, Edvardson S, Elpeleg O, Chisholm E, Juusola J, Douglas G, Guillen Sacoto MJ, Siquier-Pernet K, Saadi A, Bole-Feysot C, Nitschke P, Narravula A, Walke M, Horner MB, Day-Salvatore DL, Jayakar P, Vergano SA, Tarnopolsky MA, Hegde M, Colleaux L, Crino P, Hakonarson H. Mutations in TBCK, encoding TBC1-domain-containing kinase, lead to a recognizable syndrome of intellectual disability and hypotonia. Am J Hum Genet. 2016;98:782–788. doi: 10.1016/j.ajhg.2016.03.016. - DOI - PMC - PubMed
-
- Bidinosti M, Botta P, Kruttner S, Proenca CC, Stoehr N, Bernhard M, Fruh I, Mueller M, Bonenfant D, Voshol H, Carbone W, Neal SJ, McTighe SM, Roma G, Dolmetsch RE, Porter JA, Caroni P, Bouwmeester T, Luthi A, Galimberti I. CLK2 inhibition ameliorates autistic features associated with SHANK3 deficiency. Science. 2016;351:1199–1203. doi: 10.1126/science.aad5487. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
