

Low metformin causes a more oxidized mitochondrial NADH/NAD redox state in hepatocytes and inhibits gluconeogenesis by a redox-independent mechanism
- PMID: 30591586
- PMCID: PMC6393620
- DOI: 10.1074/jbc.RA118.006670
Low metformin causes a more oxidized mitochondrial NADH/NAD redox state in hepatocytes and inhibits gluconeogenesis by a redox-independent mechanism
Abstract
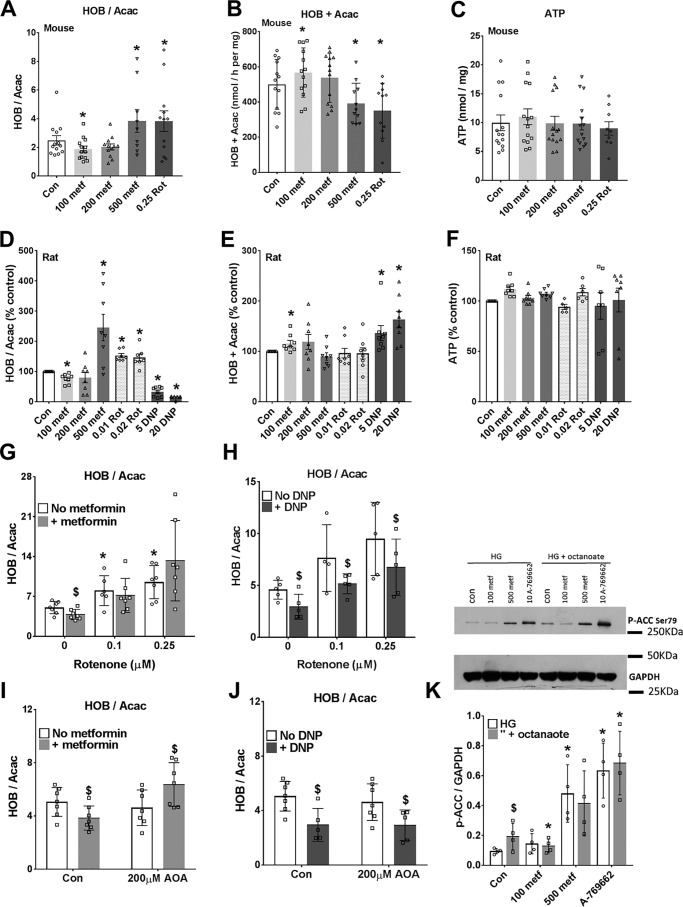
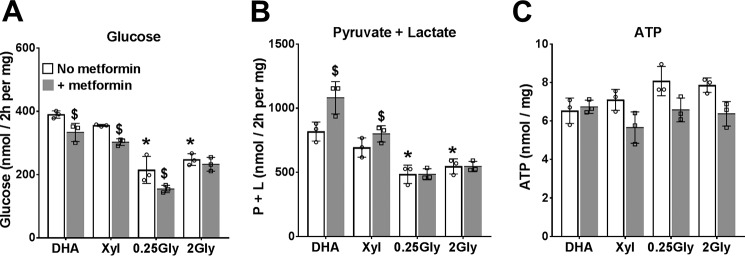
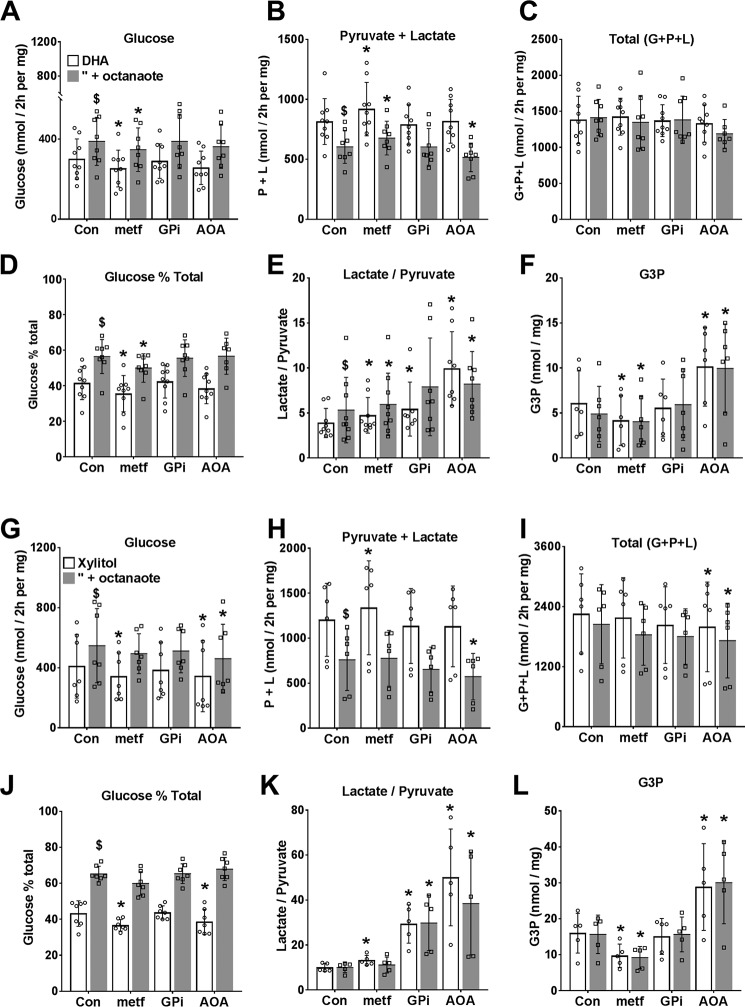
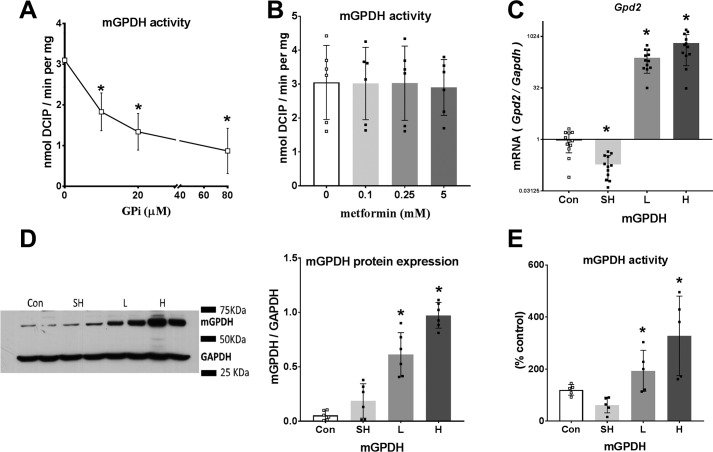
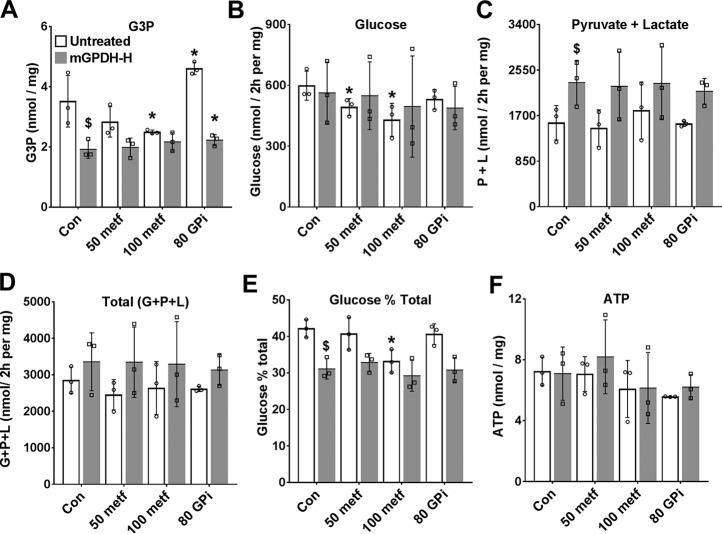
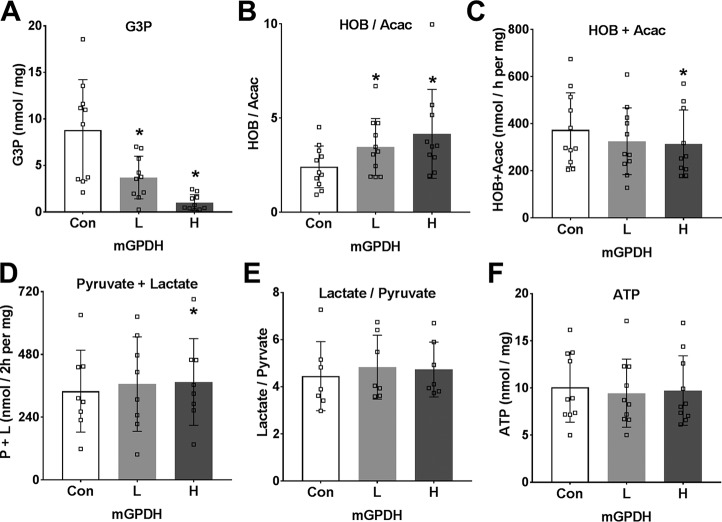
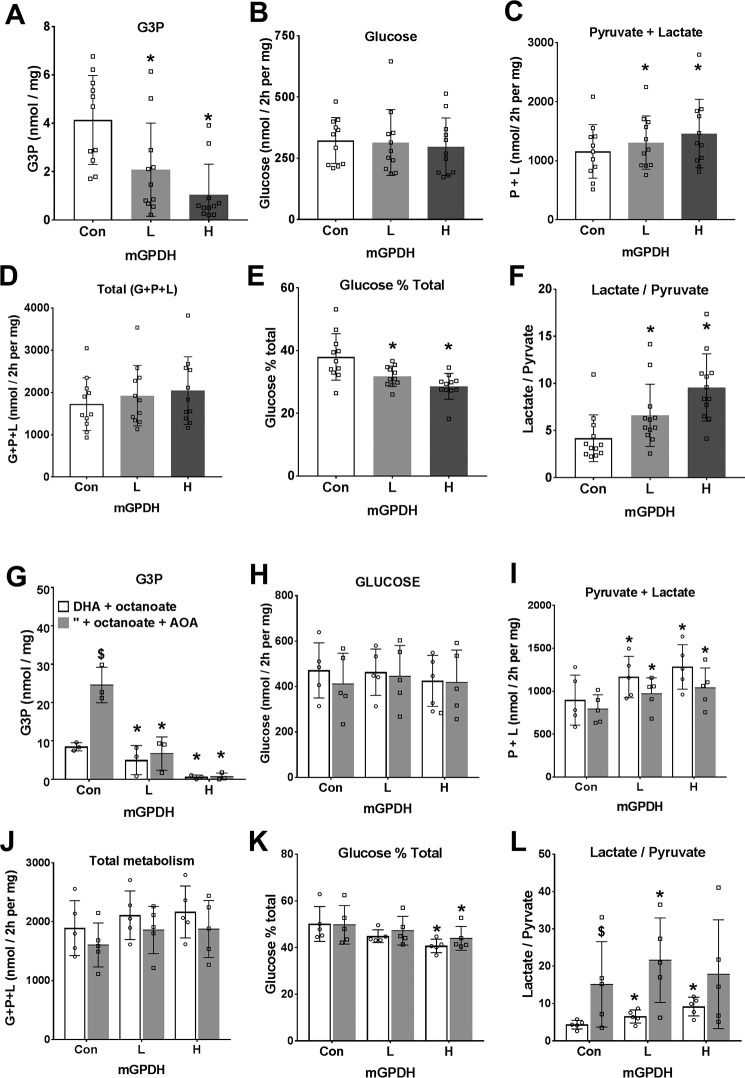
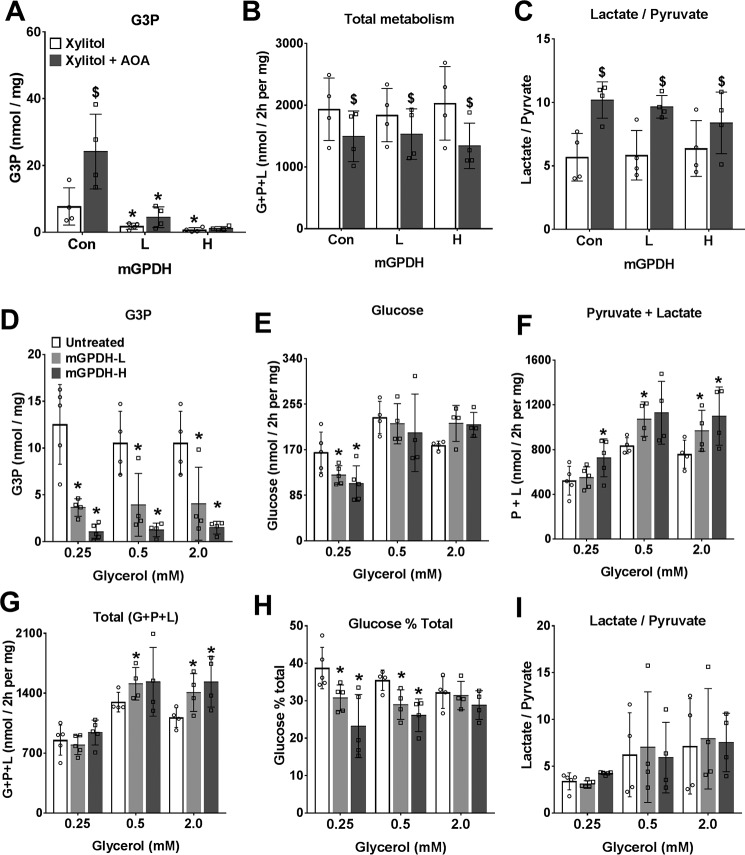
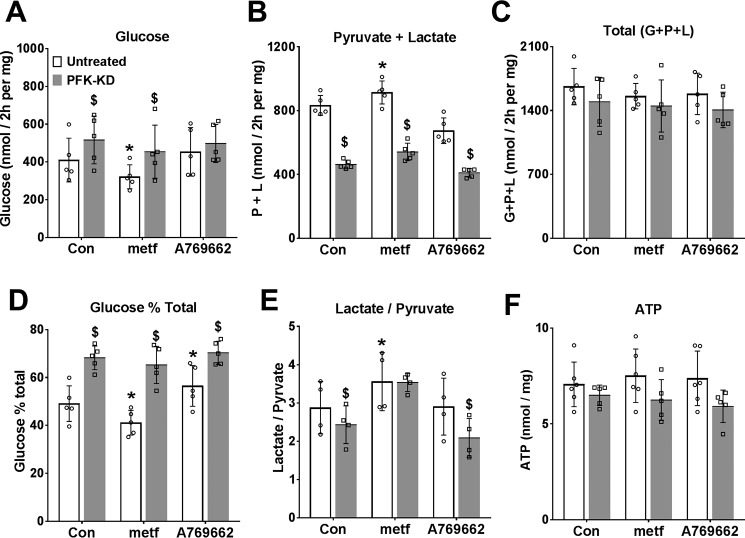
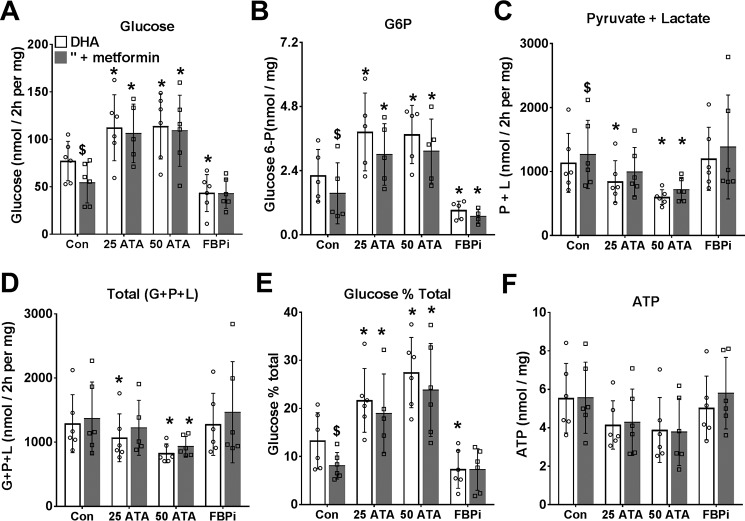
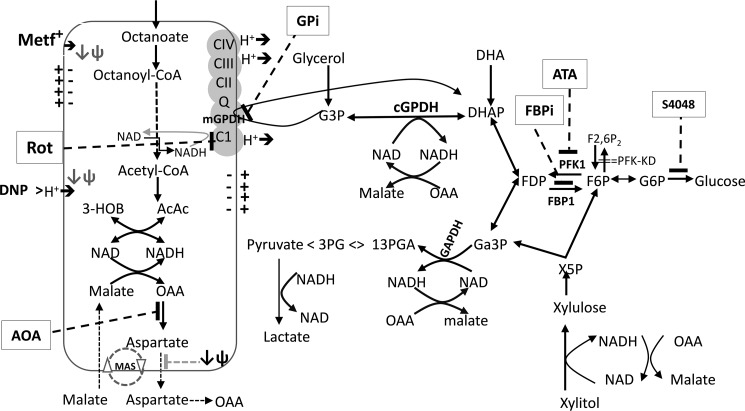
The mechanisms by which metformin (dimethylbiguanide) inhibits hepatic gluconeogenesis at concentrations relevant for type 2 diabetes therapy remain debated. Two proposed mechanisms are 1) inhibition of mitochondrial Complex 1 with consequent compromised ATP and AMP homeostasis or 2) inhibition of mitochondrial glycerophosphate dehydrogenase (mGPDH) and thereby attenuated transfer of reducing equivalents from the cytoplasm to mitochondria, resulting in a raised lactate/pyruvate ratio and redox-dependent inhibition of gluconeogenesis from reduced but not oxidized substrates. Here, we show that metformin has a biphasic effect on the mitochondrial NADH/NAD redox state in mouse hepatocytes. A low cell dose of metformin (therapeutic equivalent: <2 nmol/mg) caused a more oxidized mitochondrial NADH/NAD state and an increase in lactate/pyruvate ratio, whereas a higher metformin dose (≥5 nmol/mg) caused a more reduced mitochondrial NADH/NAD state similar to Complex 1 inhibition by rotenone. The low metformin dose inhibited gluconeogenesis from both oxidized (dihydroxyacetone) and reduced (xylitol) substrates by preferential partitioning of substrate toward glycolysis by a redox-independent mechanism that is best explained by allosteric regulation at phosphofructokinase-1 (PFK1) and/or fructose 1,6-bisphosphatase (FBP1) in association with a decrease in cell glycerol 3-phosphate, an inhibitor of PFK1, rather than by inhibition of transfer of reducing equivalents. We conclude that at a low pharmacological load, the metformin effects on the lactate/pyruvate ratio and glucose production are explained by attenuation of transmitochondrial electrogenic transport mechanisms with consequent compromised malate-aspartate shuttle and changes in allosteric effectors of PFK1 and FBP1.
Keywords: metformin; metabolic regulation; metabolic disease; gluconeogenesis; redox regulation; dimethylbiguanide; mitochondrial glycerophosphate dehydrogenase; phosphofructokinase-1.
© 2019 Alshawi and Agius.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article.
Figures
References
-
- Cao J., Meng S., Chang E., Beckwith-Fickas K., Xiong L., Cole R. N., Radovick S., Wondisford F. E., and He L. (2014) Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J. Biol. Chem. 289, 20435–20446 10.1074/jbc.M114.567271 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
