

Mechanisms protecting host cells against bacterial pore-forming toxins
- PMID: 30591958
- PMCID: PMC6420883
- DOI: 10.1007/s00018-018-2992-8
Mechanisms protecting host cells against bacterial pore-forming toxins
Abstract
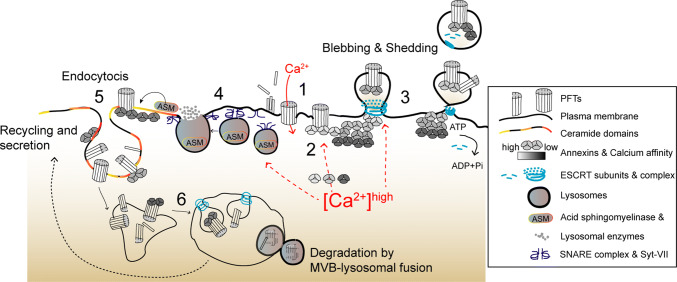
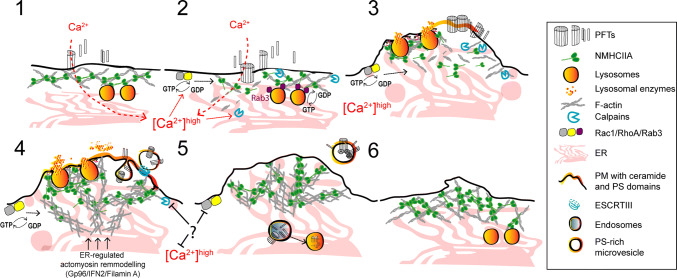
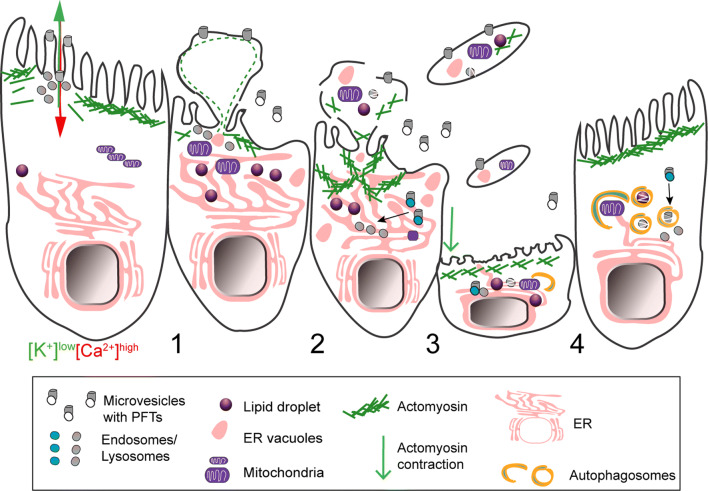
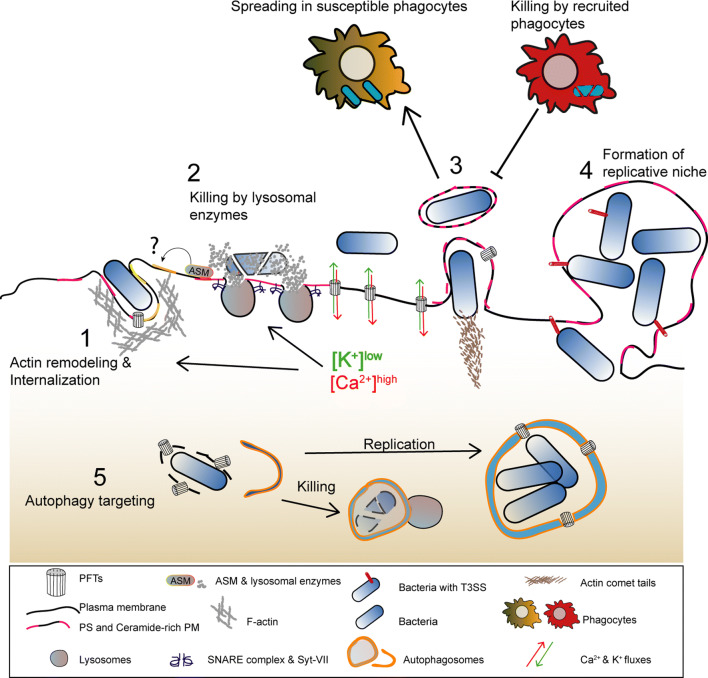
Pore-forming toxins (PFTs) are key virulence determinants produced and secreted by a variety of human bacterial pathogens. They disrupt the plasma membrane (PM) by generating stable protein pores, which allow uncontrolled exchanges between the extracellular and intracellular milieus, dramatically disturbing cellular homeostasis. In recent years, many advances were made regarding the characterization of conserved repair mechanisms that allow eukaryotic cells to recover from mechanical disruption of the PM membrane. However, the specificities of the cell recovery pathways that protect host cells against PFT-induced damage remain remarkably elusive. During bacterial infections, the coordinated action of such cell recovery processes defines the outcome of infected cells and is, thus, critical for our understanding of bacterial pathogenesis. Here, we review the cellular pathways reported to be involved in the response to bacterial PFTs and discuss their impact in single-cell recovery and infection.
Keywords: Actomyosin remodeling; Blebbing; Cholesterol-dependent cytolysins; Host signaling; Plasma membrane damage; Plasma membrane repair; Pore-forming toxins; Shedding.
Conflict of interest statement
The authors have no conflict of interest.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
