

Gene Regulatory Programs Conferring Phenotypic Identities to Human NK Cells
- PMID: 30595449
- PMCID: PMC6329660
- DOI: 10.1016/j.cell.2018.11.045
Gene Regulatory Programs Conferring Phenotypic Identities to Human NK Cells
Abstract
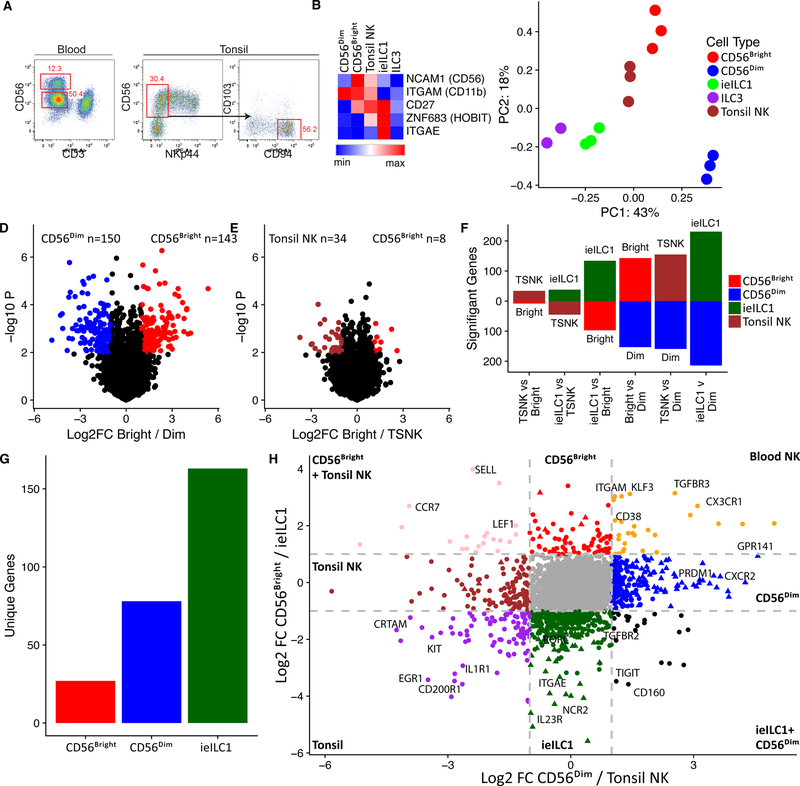
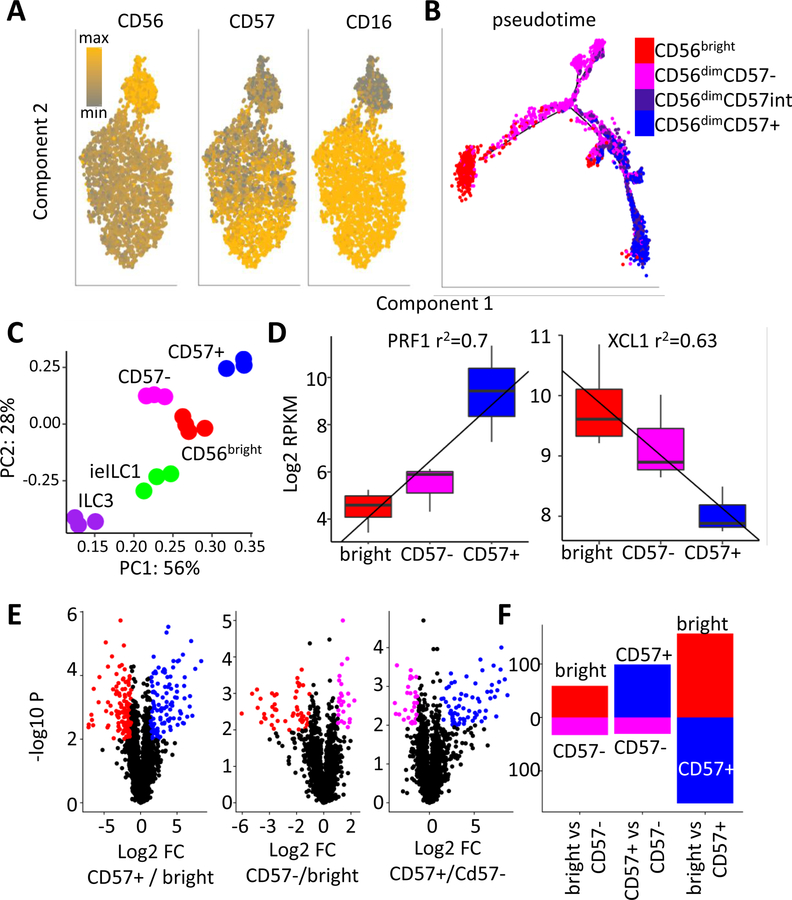
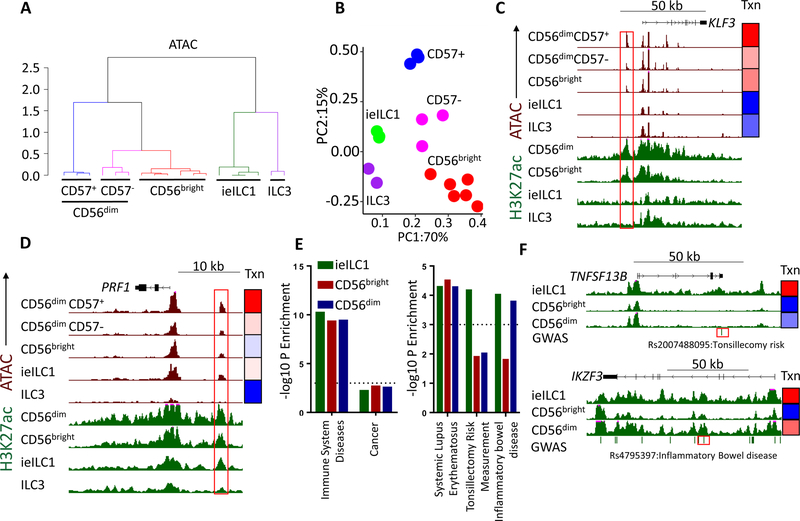
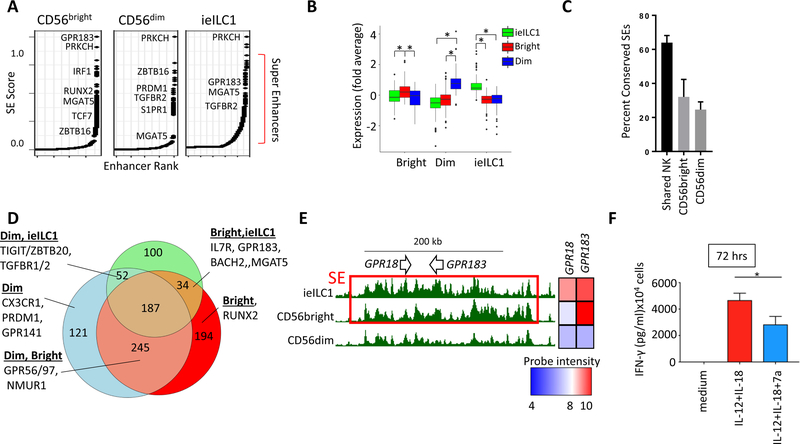
Natural killer (NK) cells develop from common progenitors but diverge into distinct subsets, which differ in cytokine production, cytotoxicity, homing, and memory traits. Given their promise in adoptive cell therapies for cancer, a deeper understanding of regulatory modules controlling clinically beneficial NK phenotypes is of high priority. We report integrated "-omics" analysis of human NK subsets, which revealed super-enhancers associated with gene cohorts that may coordinate NK functions and localization. A transcription factor-based regulatory scheme also emerged, which is evolutionarily conserved and shared by innate and adaptive lymphocytes. For both NK and T lineages, a TCF1-LEF1-MYC axis dominated the regulatory landscape of long-lived, proliferative subsets that traffic to lymph nodes. In contrast, effector populations circulating between blood and peripheral tissues shared a PRDM1-dominant landscape. This resource defines transcriptional modules, regulated by feedback loops, which may be leveraged to enhance phenotypes for NK cell-based therapies.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
References
-
- Baggio L, Laureano ÁM, Silla LM da R., and Lee DA (2017). Natural killer cell adoptive immunotherapy: Coming of age. Clin. Immunol 177, 3–11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 DE021255/DE/NIDCR NIH HHS/United States
- R21 AI120606/AI/NIAID NIH HHS/United States
- R01 AI118852/AI/NIAID NIH HHS/United States
- P51 OD011132/OD/NIH HHS/United States
- P01 AI106697/AI/NIAID NIH HHS/United States
- R56 AI134035/AI/NIAID NIH HHS/United States
- R37 AI118852/AI/NIAID NIH HHS/United States
- UL1 TR000448/TR/NCATS NIH HHS/United States
- P30 CA016058/CA/NCI NIH HHS/United States
- UL1 TR002345/TR/NCATS NIH HHS/United States
- U01 AI095542/AI/NIAID NIH HHS/United States
- R01 AI134035/AI/NIAID NIH HHS/United States
- R01 CA188286/CA/NCI NIH HHS/United States
- R01 CA176695/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
