

Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression
- PMID: 30595505
- PMCID: PMC6570409
- DOI: 10.1016/j.ccell.2018.11.015
Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression
Abstract
Diffuse intrinsic pontine gliomas (DIPGs) are incurable childhood brainstem tumors with frequent histone H3 K27M mutations and recurrent alterations in PDGFRA and TP53. We generated genetically engineered inducible mice and showed that H3.3 K27M enhanced neural stem cell self-renewal while preserving regional identity. Neonatal induction of H3.3 K27M cooperated with activating platelet-derived growth factor receptor α (PDGFRα) mutant and Trp53 loss to accelerate development of diffuse brainstem gliomas that recapitulated human DIPG gene expression signatures and showed global changes in H3K27 posttranslational modifications, but relatively restricted gene expression changes. Genes upregulated in H3.3 K27M tumors were enriched for those associated with neural development where H3K27me3 loss released the poised state of apparently bivalent promoters, whereas downregulated genes were enriched for those encoding homeodomain transcription factors.
Keywords: DIPG; H3K27me3; PDGFRA; bivalent; epigenetic; glioma; histone H3 K27M; knockin; mouse; oncohistone.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures
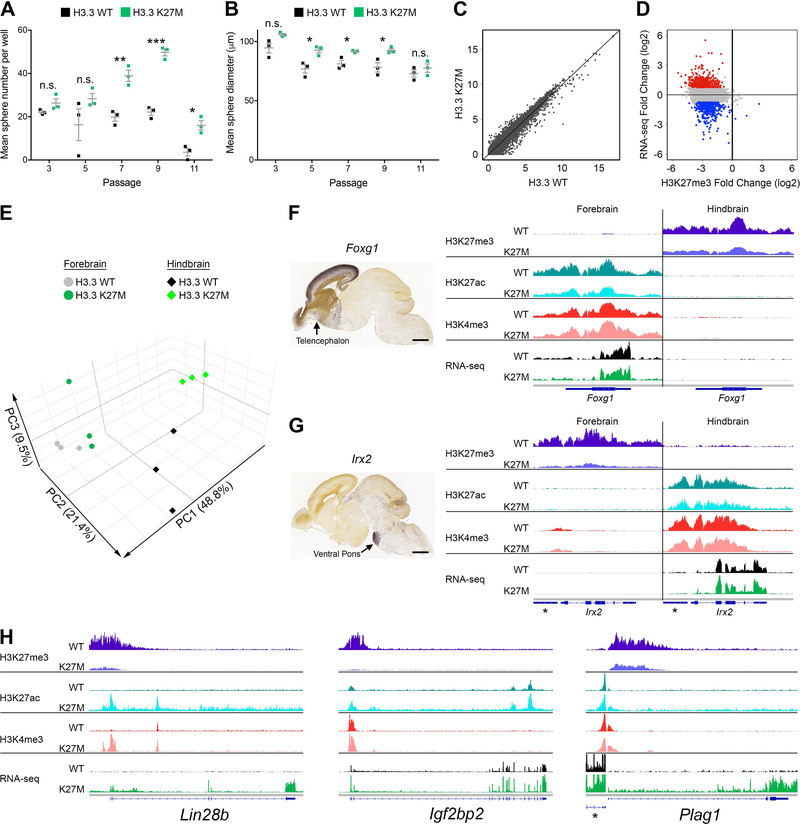
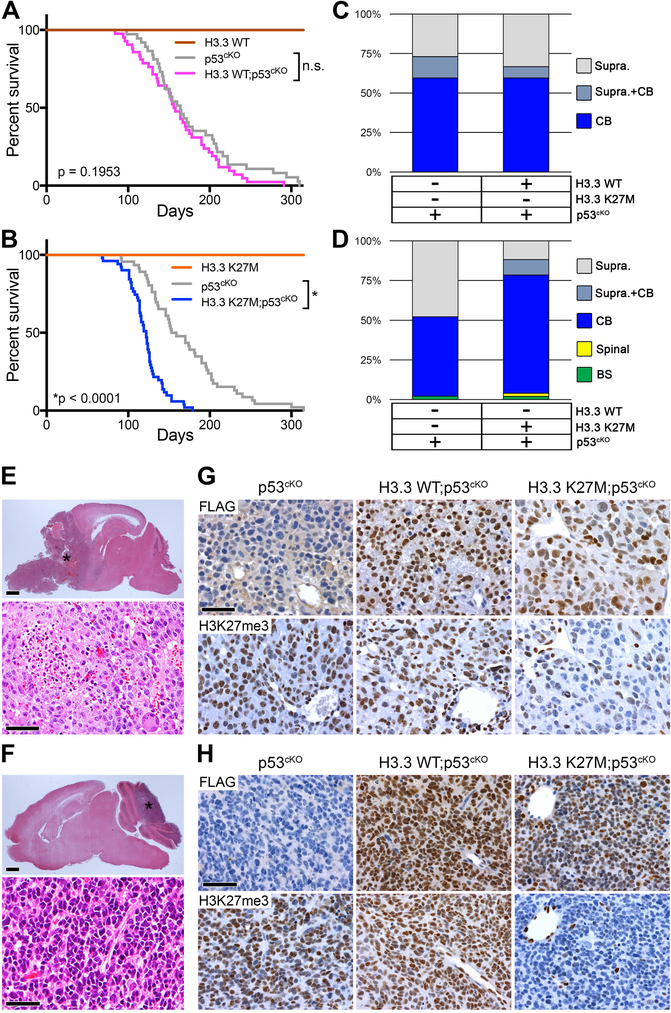
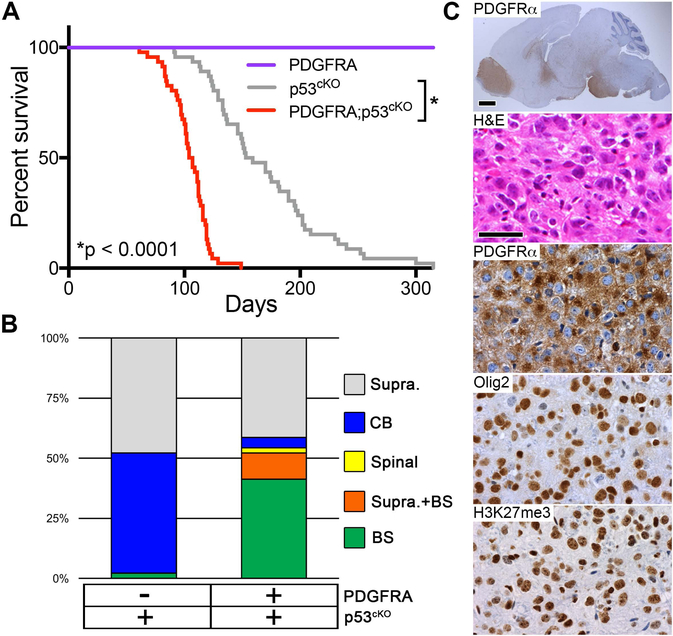
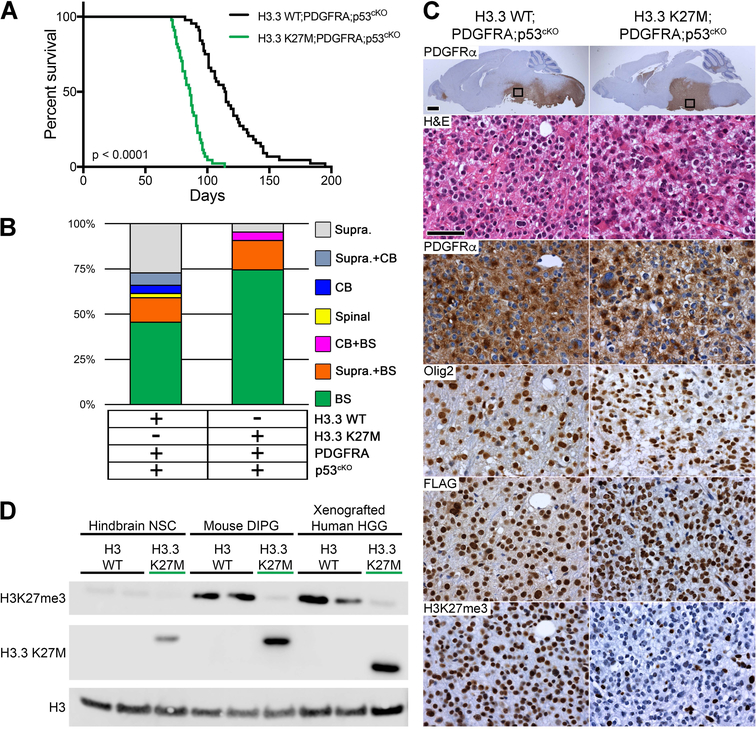
Comment in
-
Tails of a Super Histone.Cancer Cell. 2019 Jan 14;35(1):7-9. doi: 10.1016/j.ccell.2018.12.005. Cancer Cell. 2019. PMID: 30645977
References
-
- Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al. (2013). Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672. - PubMed
-
- Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U, et al. (2014). Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46, 451–456. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
