

Bayesian multiple logistic regression for case-control GWAS
- PMID: 30596640
- PMCID: PMC6329526
- DOI: 10.1371/journal.pgen.1007856
Bayesian multiple logistic regression for case-control GWAS
Abstract
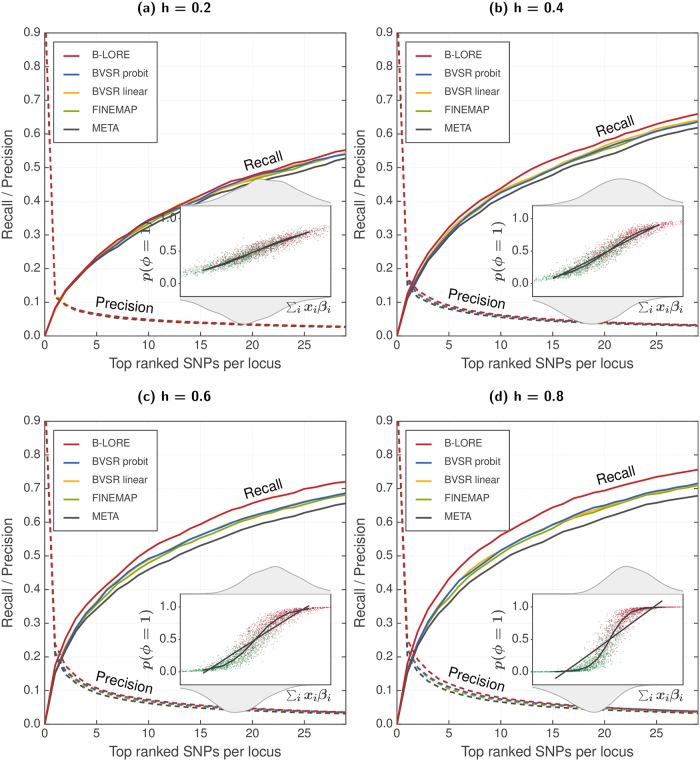
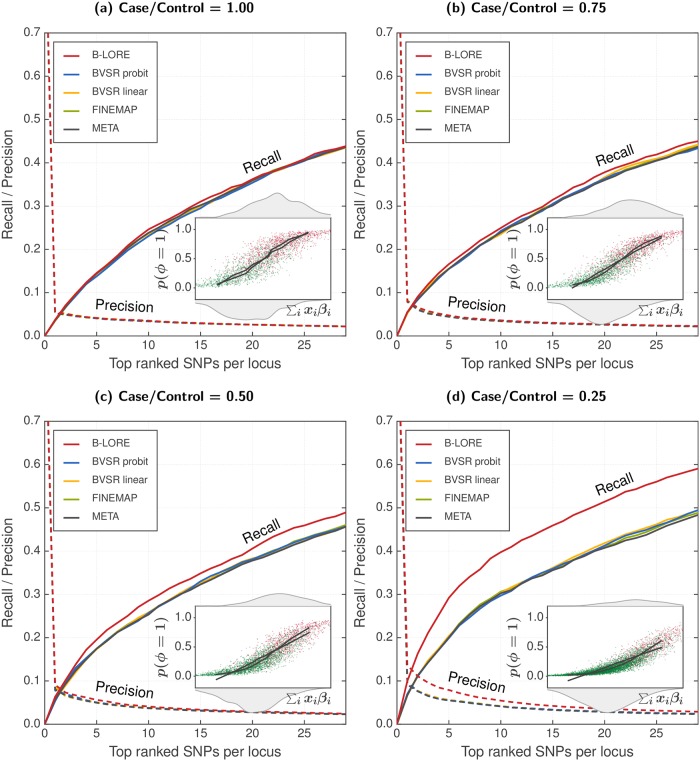
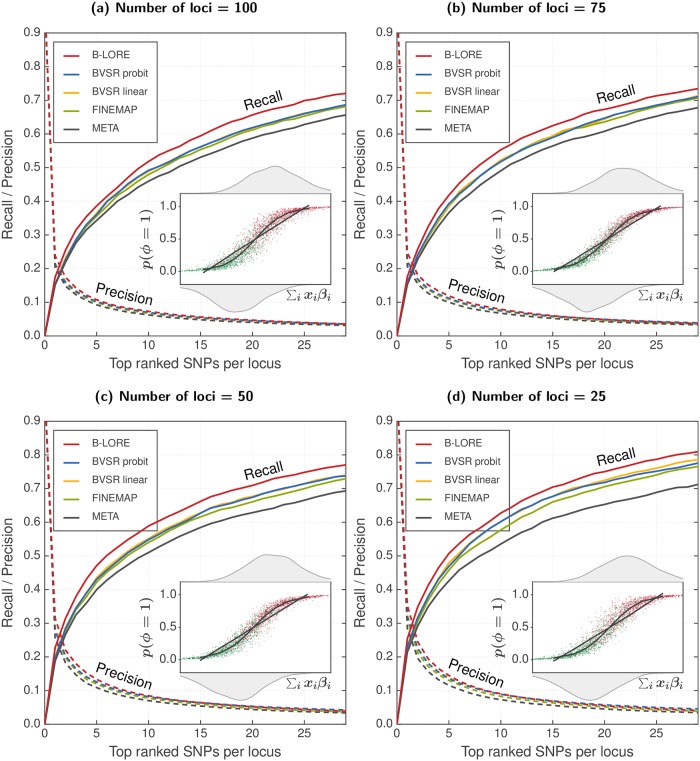
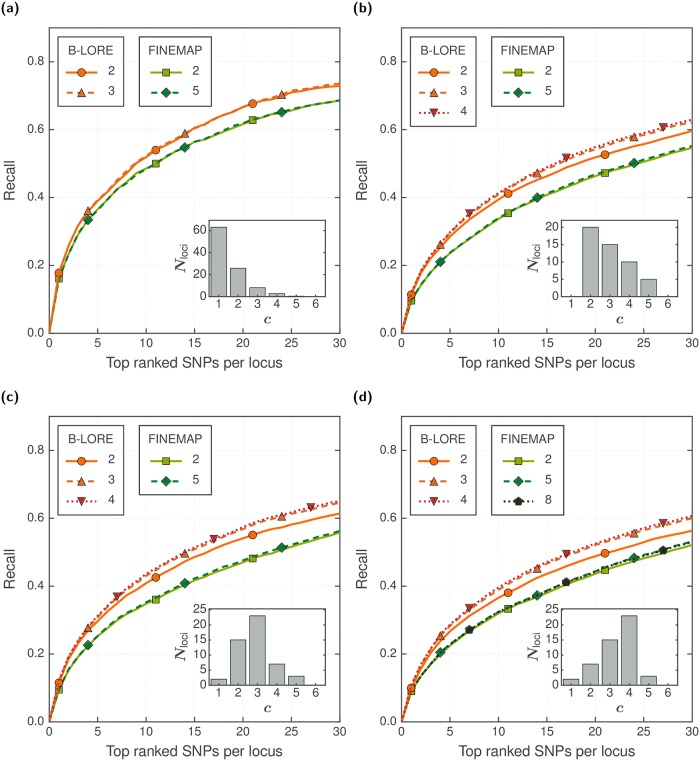
Genetic variants in genome-wide association studies (GWAS) are tested for disease association mostly using simple regression, one variant at a time. Standard approaches to improve power in detecting disease-associated SNPs use multiple regression with Bayesian variable selection in which a sparsity-enforcing prior on effect sizes is used to avoid overtraining and all effect sizes are integrated out for posterior inference. For binary traits, the logistic model has not yielded clear improvements over the linear model. For multi-SNP analysis, the logistic model required costly and technically challenging MCMC sampling to perform the integration. Here, we introduce the quasi-Laplace approximation to solve the integral and avoid MCMC sampling. We expect the logistic model to perform much better than multiple linear regression except when predicted disease risks are spread closely around 0.5, because only close to its inflection point can the logistic function be well approximated by a linear function. Indeed, in extensive benchmarks with simulated phenotypes and real genotypes, our Bayesian multiple LOgistic REgression method (B-LORE) showed considerable improvements (1) when regressing on many variants in multiple loci at heritabilities ≥ 0.4 and (2) for unbalanced case-control ratios. B-LORE also enables meta-analysis by approximating the likelihood functions of individual studies by multivariate normal distributions, using their means and covariance matrices as summary statistics. Our work should make sparse multiple logistic regression attractive also for other applications with binary target variables. B-LORE is freely available from: https://github.com/soedinglab/b-lore.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
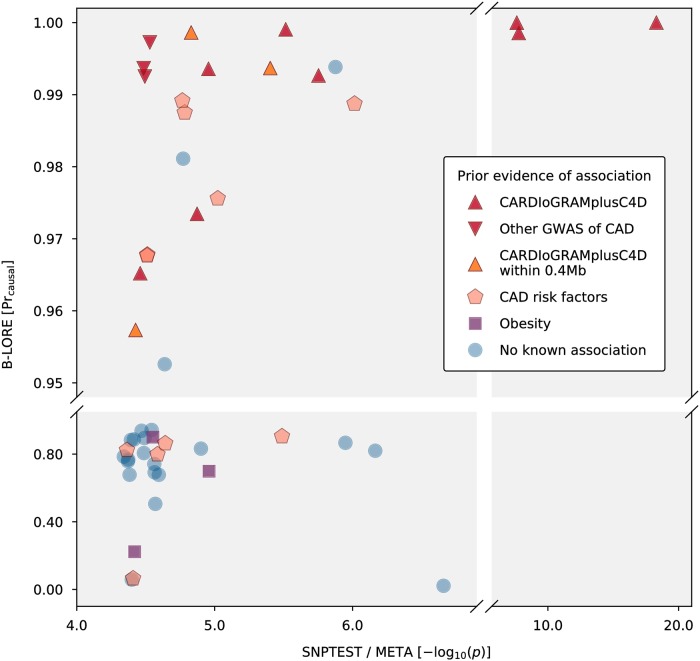
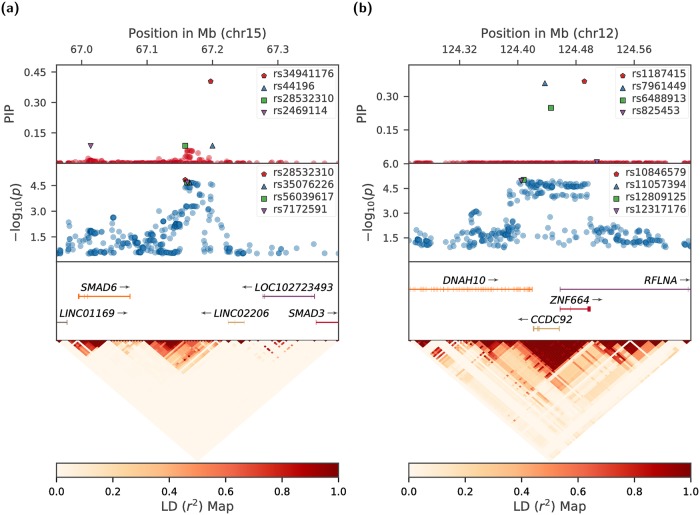
References
-
- Guan Y, Stephens M. Bayesian variable selection regression for genome-wide association studies and other large-scale problems. Annals of Applied Statistics. 2011;5(3):1780–1815. 10.1214/11-AOAS455 - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
