

MoMo: discovery of statistically significant post-translational modification motifs
- PMID: 30596994
- PMCID: PMC6691336
- DOI: 10.1093/bioinformatics/bty1058
MoMo: discovery of statistically significant post-translational modification motifs
Abstract
Motivation: Post-translational modifications (PTMs) of proteins are associated with many significant biological functions and can be identified in high throughput using tandem mass spectrometry. Many PTMs are associated with short sequence patterns called 'motifs' that help localize the modifying enzyme. Accordingly, many algorithms have been designed to identify these motifs from mass spectrometry data. Accurate statistical confidence estimates for discovered motifs are critically important for proper interpretation and in the design of downstream experimental validation.
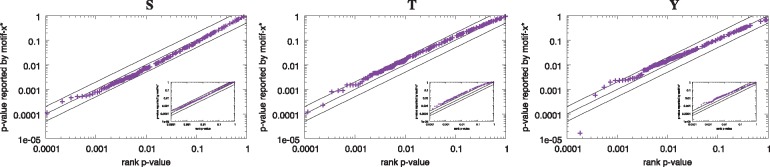
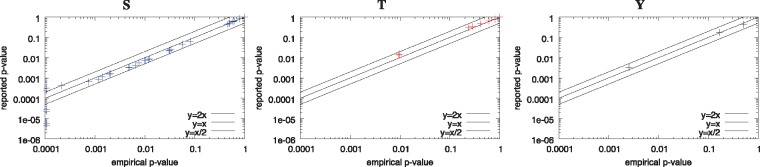
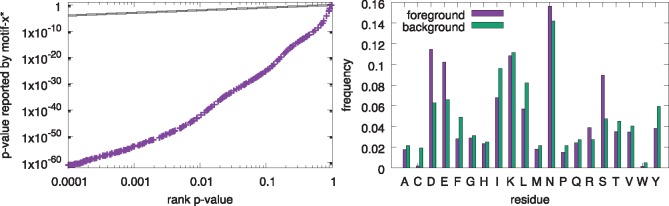
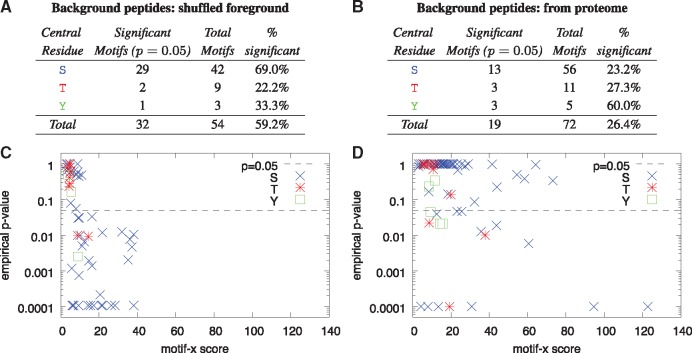
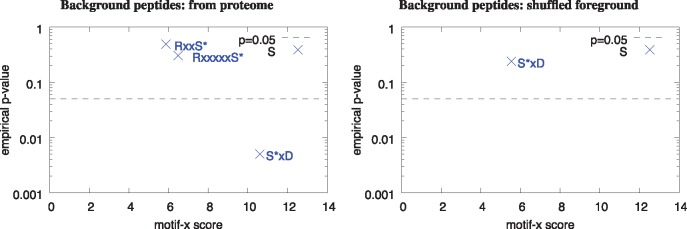
Results: We describe a method for assigning statistical confidence estimates to PTM motifs, and we demonstrate that this method provides accurate P-values on both simulated and real data. Our methods are implemented in MoMo, a software tool for discovering motifs among sets of PTMs that we make available as a web server and as downloadable source code. MoMo re-implements the two most widely used PTM motif discovery algorithms-motif-x and MoDL-while offering many enhancements. Relative to motif-x, MoMo offers improved statistical confidence estimates and more accurate calculation of motif scores. The MoMo web server offers more proteome databases, more input formats, larger inputs and longer running times than the motif-x web server. Finally, our study demonstrates that the confidence estimates produced by motif-x are inaccurate. This inaccuracy stems in part from the common practice of drawing 'background' peptides from an unshuffled proteome database. Our results thus suggest that many of the papers that use motif-x to find motifs may be reporting results that lack statistical support.
Availability and implementation: The MoMo web server and source code are provided at http://meme-suite.org.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2018. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
Similar articles
-
STREME: accurate and versatile sequence motif discovery.Bioinformatics. 2021 Sep 29;37(18):2834-2840. doi: 10.1093/bioinformatics/btab203. Bioinformatics. 2021. PMID: 33760053 Free PMC article.
-
Non-parametric Bayesian approach to post-translational modification refinement of predictions from tandem mass spectrometry.Bioinformatics. 2013 Apr 1;29(7):821-9. doi: 10.1093/bioinformatics/btt056. Epub 2013 Feb 17. Bioinformatics. 2013. PMID: 23419374
-
MEME SUITE: tools for motif discovery and searching.Nucleic Acids Res. 2009 Jul;37(Web Server issue):W202-8. doi: 10.1093/nar/gkp335. Epub 2009 May 20. Nucleic Acids Res. 2009. PMID: 19458158 Free PMC article.
-
Software eyes for protein post-translational modifications.Mass Spectrom Rev. 2015 Mar-Apr;34(2):133-47. doi: 10.1002/mas.21425. Epub 2014 Jun 2. Mass Spectrom Rev. 2015. PMID: 24889695 Review.
-
Bioinformatics Approaches for Predicting Disordered Protein Motifs.Adv Exp Med Biol. 2015;870:291-318. doi: 10.1007/978-3-319-20164-1_9. Adv Exp Med Biol. 2015. PMID: 26387106 Review.
Cited by
-
HypDB: A functionally annotated web-based database of the proline hydroxylation proteome.PLoS Biol. 2022 Aug 26;20(8):e3001757. doi: 10.1371/journal.pbio.3001757. eCollection 2022 Aug. PLoS Biol. 2022. PMID: 36026437 Free PMC article.
-
Dihydroartemisinin regulates immune cell heterogeneity by triggering a cascade reaction of CDK and MAPK phosphorylation.Signal Transduct Target Ther. 2022 Jul 11;7(1):222. doi: 10.1038/s41392-022-01028-5. Signal Transduct Target Ther. 2022. PMID: 35811310 Free PMC article.
-
Multiple Layers of Phospho-Regulation Coordinate Metabolism and the Cell Cycle in Budding Yeast.Front Cell Dev Biol. 2019 Dec 17;7:338. doi: 10.3389/fcell.2019.00338. eCollection 2019. Front Cell Dev Biol. 2019. PMID: 31921850 Free PMC article.
-
Robust unsupervised deconvolution of linear motifs characterizes 68 protein modifications at proteome scale.Sci Rep. 2021 Nov 18;11(1):22490. doi: 10.1038/s41598-021-01971-3. Sci Rep. 2021. PMID: 34795380 Free PMC article.
-
Phosphorylation of multiple proteins involved in ciliogenesis by Tau Tubulin kinase 2.Mol Biol Cell. 2020 May 1;31(10):1032-1046. doi: 10.1091/mbc.E19-06-0334. Epub 2020 Mar 4. Mol Biol Cell. 2020. PMID: 32129703 Free PMC article.
References
-
- Bailey T.L., Elkan C. (1995) The value of prior knowledge in discovering motifs with MEME. In: Proceedings of the Third International Conference on Intelligent Systems for Molecular Biology, Cambridge, United Kingdom, July 16-19, 1995, pp. 21–29. - PubMed
-
- Chou M.F., Schwartz D. (2011) Biological sequence motif discovery using motif-x. Curr. Protocols Bioinform., 35, 13.15.1–13.15.24. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
