

Revealing transcription factor and histone modification co-localization and dynamics across cell lines by integrating ChIP-seq and RNA-seq data
- PMID: 30598100
- PMCID: PMC6311957
- DOI: 10.1186/s12864-018-5278-5
Revealing transcription factor and histone modification co-localization and dynamics across cell lines by integrating ChIP-seq and RNA-seq data
Abstract
Background: Interactions among transcription factors (TFs) and histone modifications (HMs) play an important role in the precise regulation of gene expression. The context specificity of those interactions and further its dynamics in normal and disease remains largely unknown. Recent development in genomics technology enables transcription profiling by RNA-seq and protein's binding profiling by ChIP-seq. Integrative analysis of the two types of data allows us to investigate TFs and HMs interactions both from the genome co-localization and downstream target gene expression.
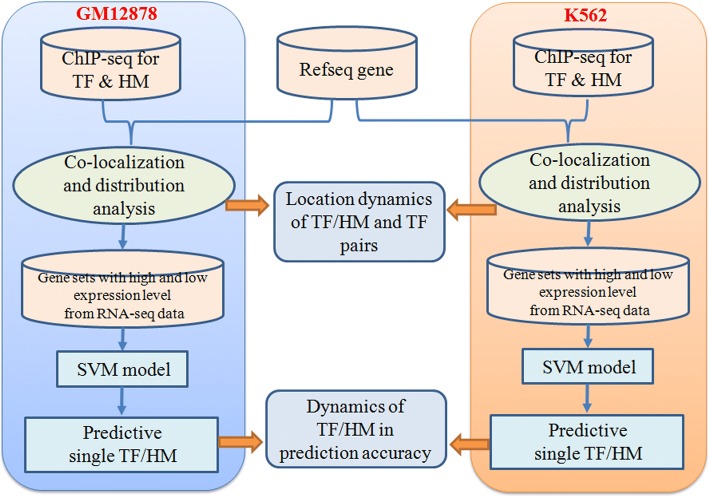
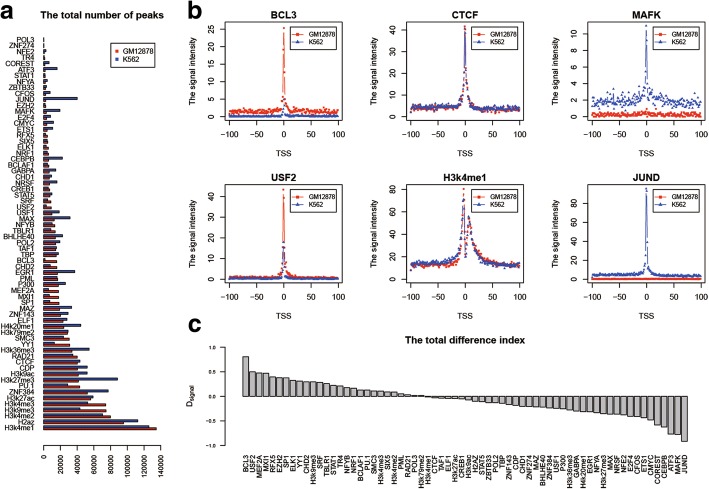
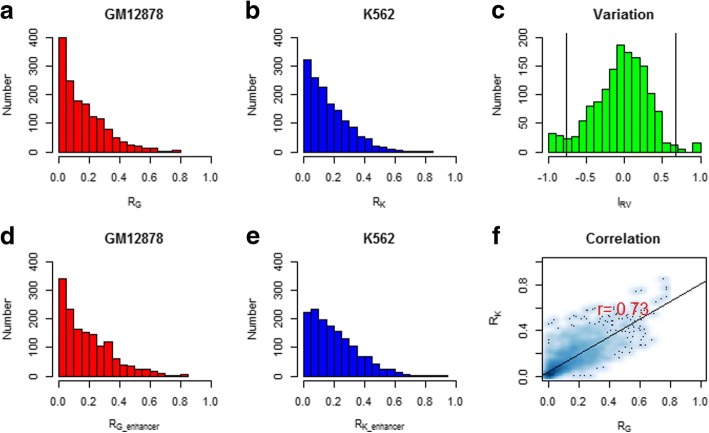
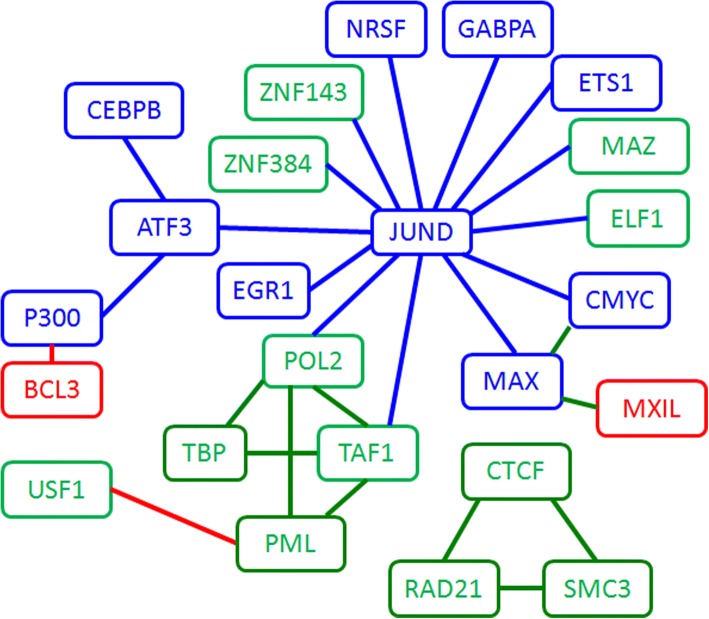
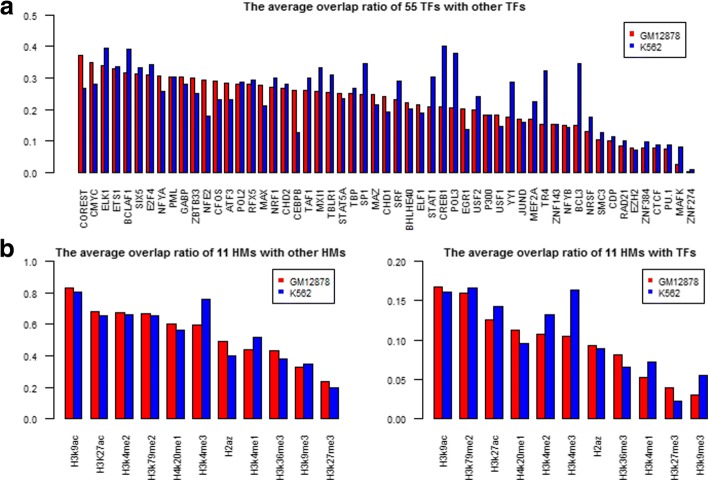
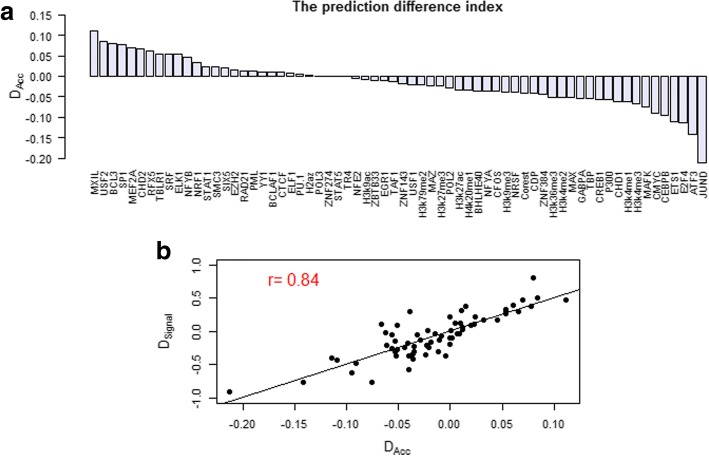
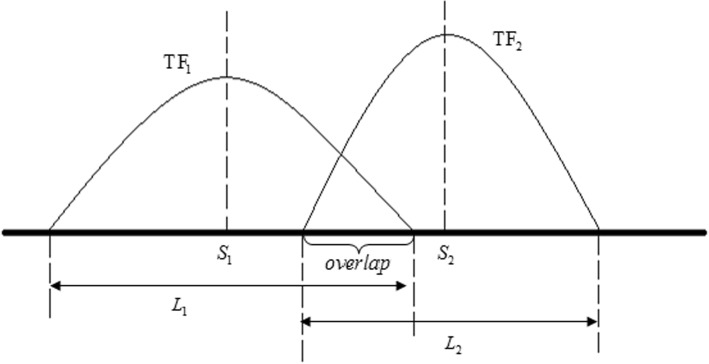
Results: We propose a integrative pipeline to explore the co-localization of 55 TFs and 11 HMs and its dynamics in human GM12878 and K562 by matched ChIP-seq and RNA-seq data from ENCODE. We classify TFs and HMs into three types based on their binding enrichment around transcription start site (TSS). Then a set of statistical indexes are proposed to characterize the TF-TF and TF-HM co-localizations. We found that Rad21, SMC3, and CTCF co-localized across five cell lines. High resolution Hi-C data in GM12878 shows that they associate most of the Hi-C peak loci with a specific CTCF-motif "anchor" and supports that CTCF, SMC3, and RAD2 co-localization serves important role in 3D chromatin structure. Meanwhile, 17 TF-TF pairs are highly dynamic between GM12878 and K562. We then build SVM models to correlate high and low expression level of target genes with TF binding and HM strength. We found that H3k9ac, H3k27ac, and three TFs (ELF1, TAF1, and POL2) are predictive with the accuracy about 85~92%.
Conclusion: We propose a pipeline to analyze the co-localization of TF and HM and their dynamics across cell lines from ChIP-seq, and investigate their regulatory potency by RNA-seq. The integrative analysis of two level data reveals new insight for the cooperation of TFs and HMs and is helpful in understanding cell line specificity of TF/HM interactions.
Keywords: Co-localization; Dynamics; Histone modification; Transcription factor.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
